

Cholesterol 25-hydroxylase promotes efferocytosis and resolution of lung inflammation
- PMID: 32343675
- PMCID: PMC7308063
- DOI: 10.1172/jci.insight.137189
Cholesterol 25-hydroxylase promotes efferocytosis and resolution of lung inflammation
Abstract
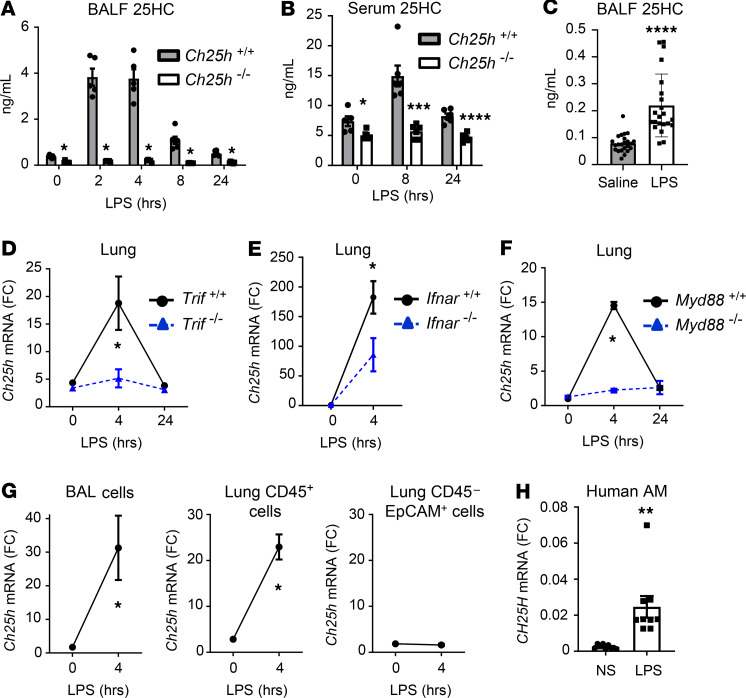
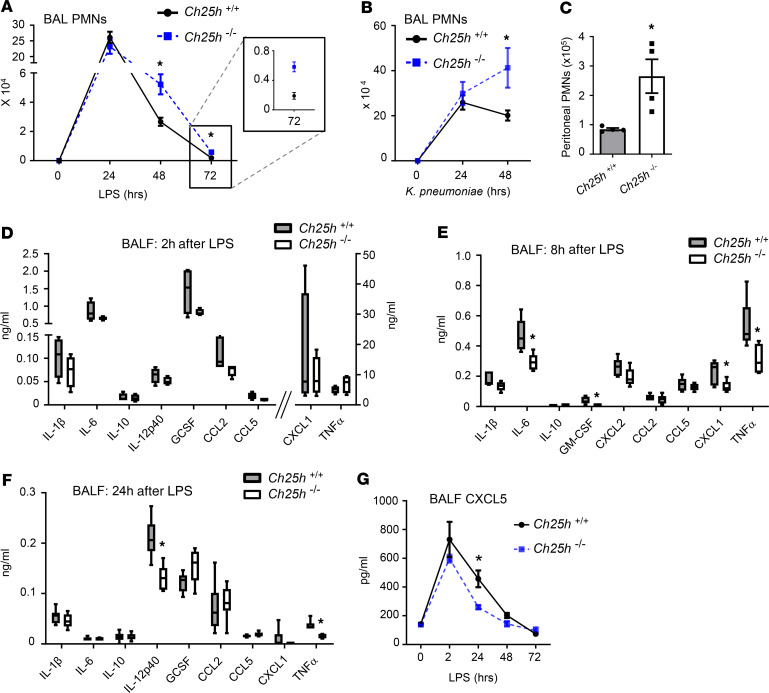
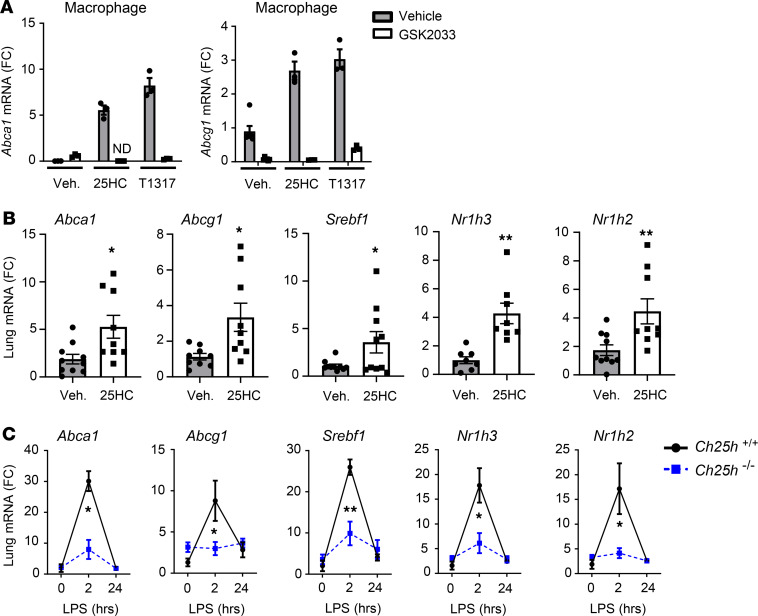
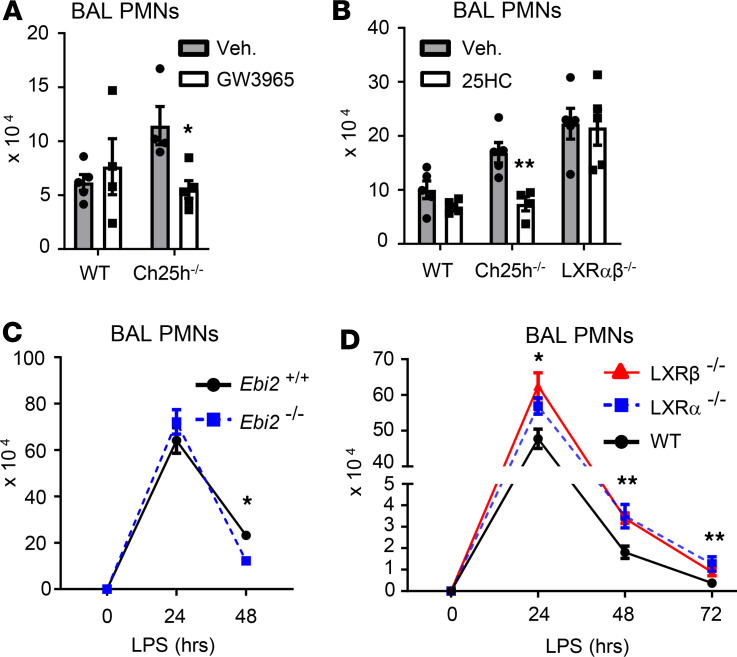
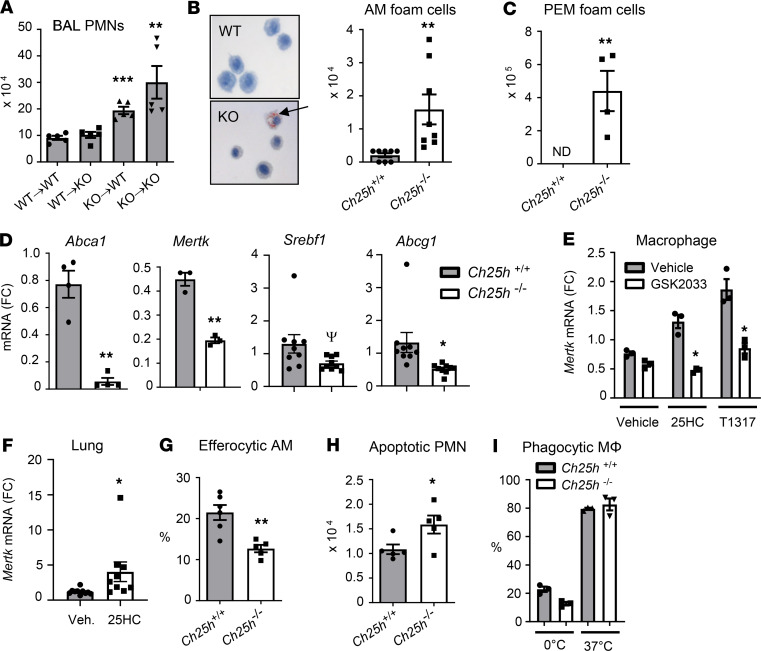
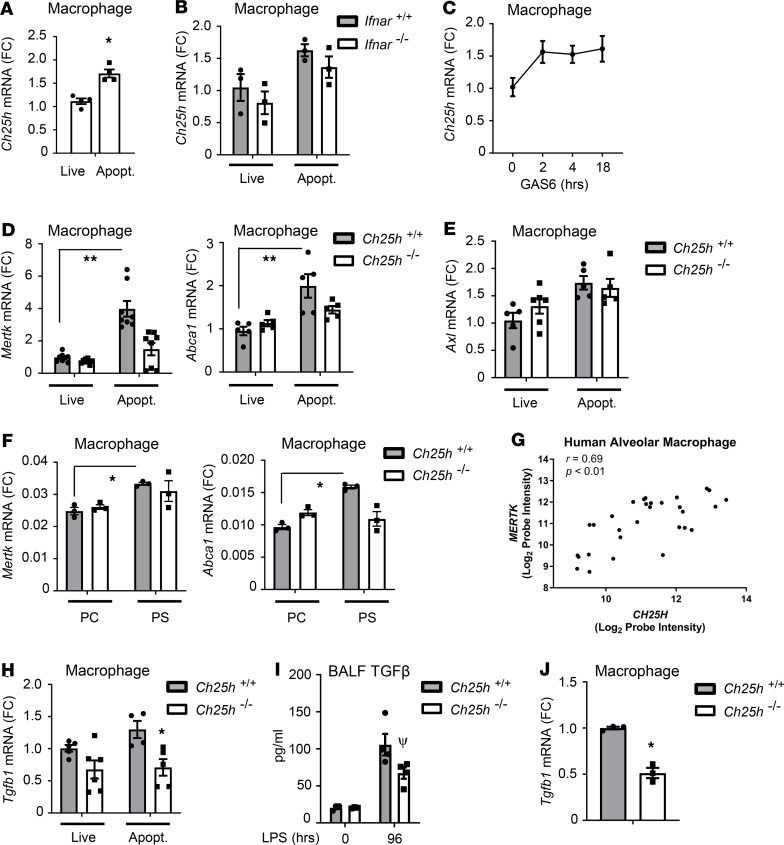
Alveolar macrophages (AM) play a central role in initiation and resolution of lung inflammation, but the integration of these opposing core functions is poorly understood. AM expression of cholesterol 25-hydroxylase (CH25H), the primary biosynthetic enzyme for 25-hydroxycholesterol (25HC), far exceeds the expression of macrophages in other tissues, but no role for CH25H has been defined in lung biology. As 25HC is an agonist for the antiinflammatory nuclear receptor, liver X receptor (LXR), we speculated that CH25H might regulate inflammatory homeostasis in the lung. Here, we show that, of natural oxysterols or sterols, 25HC is induced in the inflamed lung of mice and humans. Ch25h-/- mice fail to induce 25HC and LXR target genes in the lung after LPS inhalation and exhibit delayed resolution of airway neutrophilia, which can be rescued by systemic treatment with either 25HC or synthetic LXR agonists. LXR-null mice also display delayed resolution, suggesting that native oxysterols promote resolution. During resolution, Ch25h is induced in macrophages upon their encounter with apoptotic cells and is required for LXR-dependent prevention of AM lipid overload, induction of Mertk, efferocytic resolution of airway neutrophilia, and induction of TGF-β. CH25H/25HC/LXR is, thus, an inducible metabolic axis that programs AMs for efferocytic resolution of inflammation.
Keywords: Cholesterol; Inflammation; Innate immunity; Macrophages; Pulmonology.
Conflict of interest statement
Figures
References
-
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101(4):890–898. doi: 10.1172/JCI1112. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
