

Genetic basis and molecular biology of cardiac arrhythmias in cardiomyopathies
- PMID: 32348453
- PMCID: PMC7341170
- DOI: 10.1093/cvr/cvaa116
Genetic basis and molecular biology of cardiac arrhythmias in cardiomyopathies
Abstract
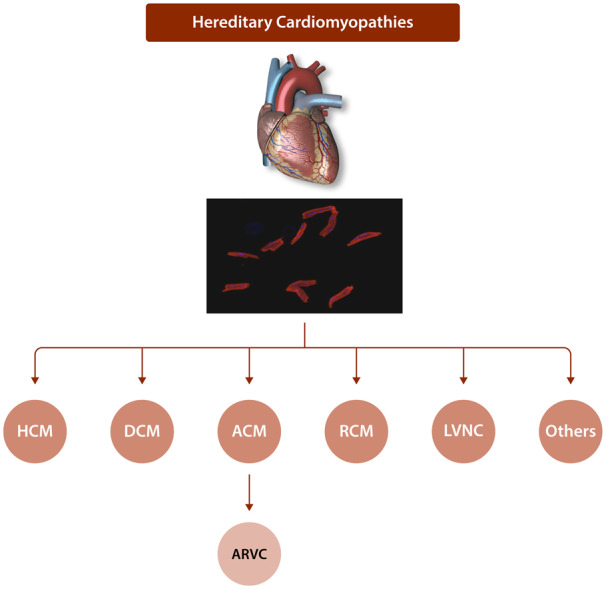
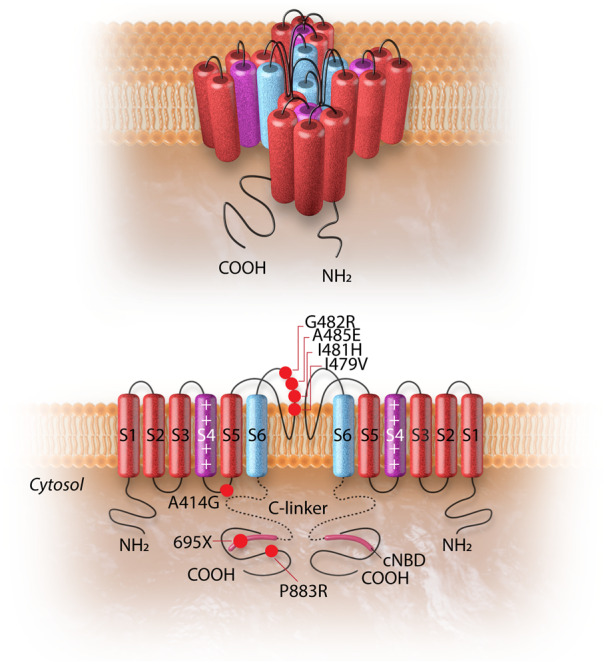
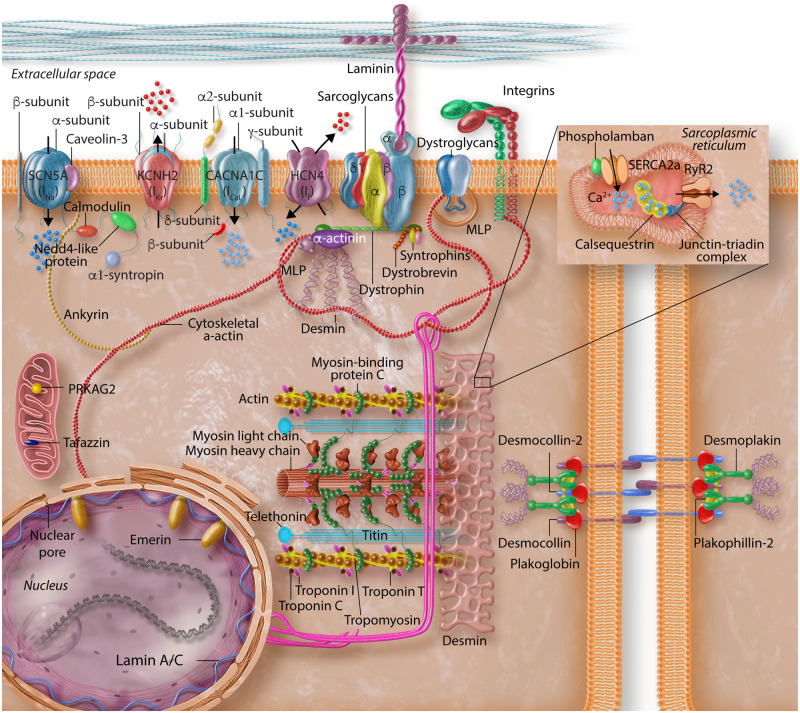
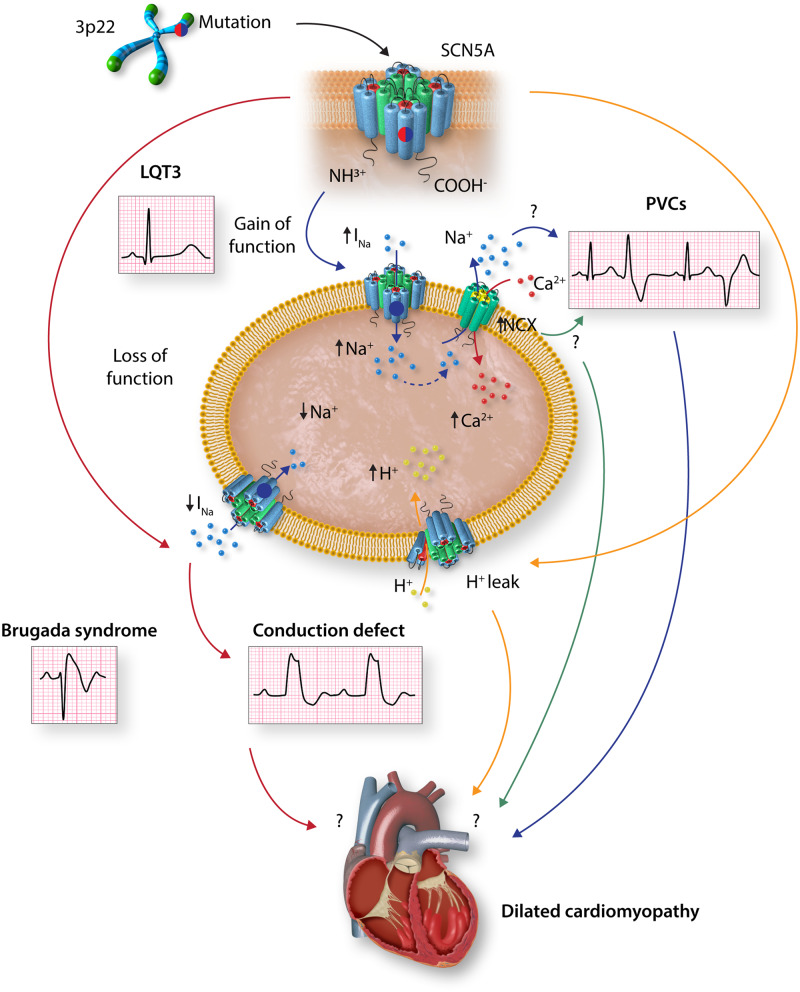
Cardiac arrhythmias are common, often the first, and sometimes the life-threatening manifestations of hereditary cardiomyopathies. Pathogenic variants in several genes known to cause hereditary cardiac arrhythmias have also been identified in the sporadic cases and small families with cardiomyopathies. These findings suggest a shared genetic aetiology of a subset of hereditary cardiomyopathies and cardiac arrhythmias. The concept of a shared genetic aetiology is in accord with the complex and exquisite interplays that exist between the ion currents and cardiac mechanical function. However, neither the causal role of cardiac arrhythmias genes in cardiomyopathies is well established nor the causal role of cardiomyopathy genes in arrhythmias. On the contrary, secondary changes in ion currents, such as post-translational modifications, are common and contributors to the pathogenesis of arrhythmias in cardiomyopathies through altering biophysical and functional properties of the ion channels. Moreover, structural changes, such as cardiac hypertrophy, dilatation, and fibrosis provide a pro-arrhythmic substrate in hereditary cardiomyopathies. Genetic basis and molecular biology of cardiac arrhythmias in hereditary cardiomyopathies are discussed.
Keywords: Genetics • Cardiomyopathy • Arrhythmias • Ion channels • Electrophysiology • Sudden death.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author(s) 2020. For permissions, please email: journals.permissions@oup.com.
Figures
References
-
- McKenna W, Maron J, Barry J, Thiene G.. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res 2017;121:722–730. - PubMed
-
- Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, Davis AM, Thompson T, Connell V, Wallace J, Naylor C, Crawford J, Love DR, Hallam L, White J, Lawrence C, Lynch M, Morgan N, James P, Du Sart D, Puranik R, Langlois N, Vohra J, Winship I, Atherton J, McGaughran J, Skinner JR, Semsarian C.. A prospective study of sudden cardiac death among children and young adults. N Engl J Med 2016;374:2441–2452. - PubMed
-
- Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO.. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980. Circulation 2009;119:1085–1092. - PubMed
-
- O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, McKenna WJ, Omar RZ, Elliott PM, Hypertrophic Cardiomyopathy Outcomes Investigators. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014;35:2010–2020. - PubMed
