

Inferring spatial and signaling relationships between cells from single cell transcriptomic data
- PMID: 32350282
- PMCID: PMC7190659
- DOI: 10.1038/s41467-020-15968-5
Inferring spatial and signaling relationships between cells from single cell transcriptomic data
Abstract
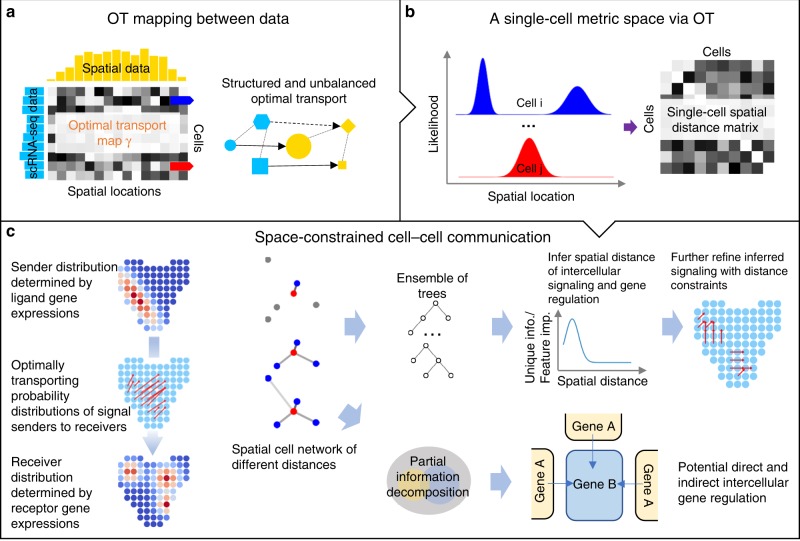
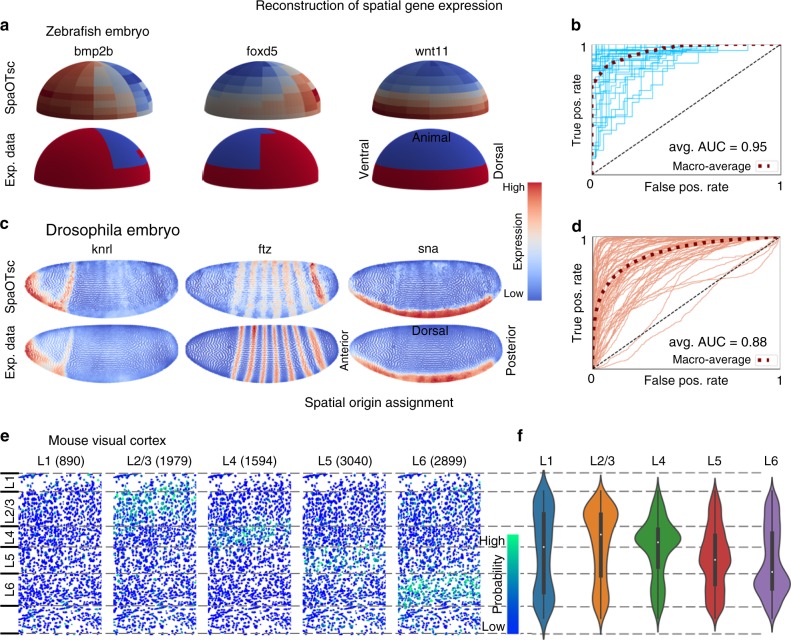
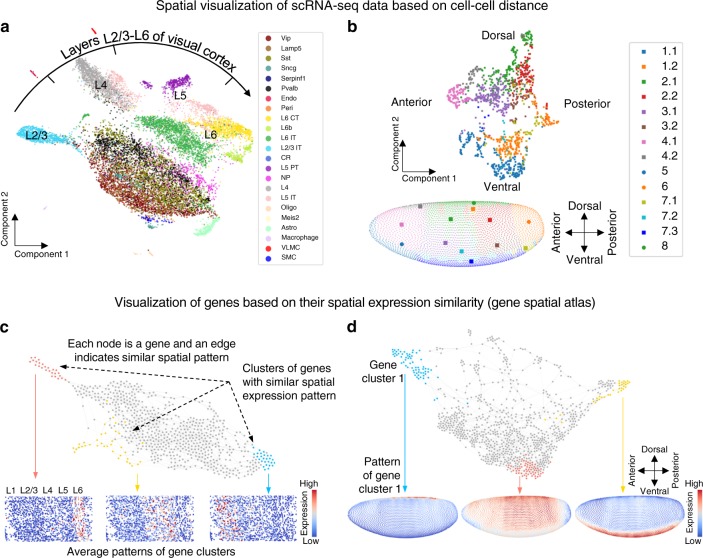
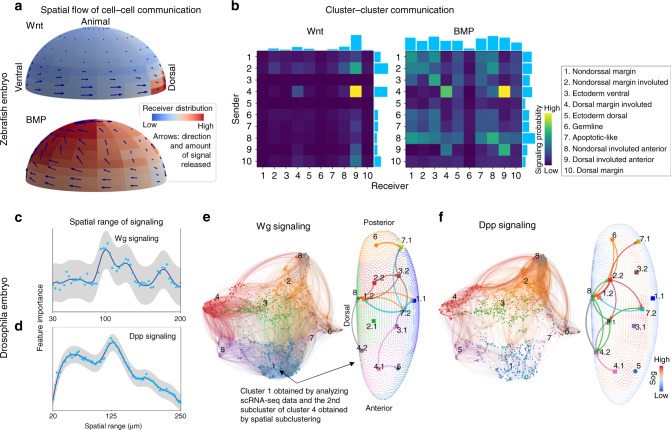
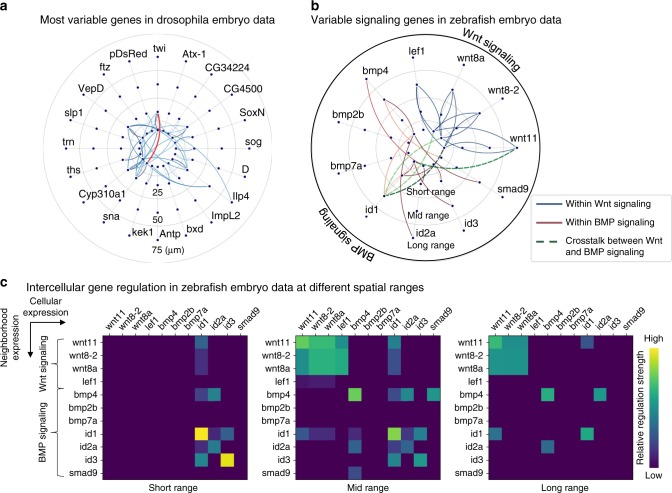
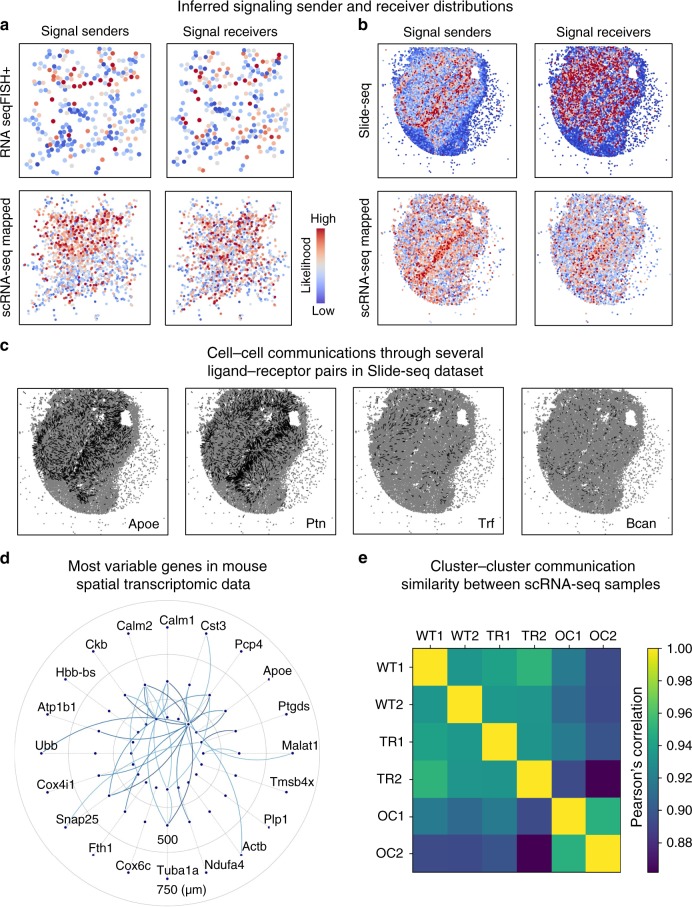
Single-cell RNA sequencing (scRNA-seq) provides details for individual cells; however, crucial spatial information is often lost. We present SpaOTsc, a method relying on structured optimal transport to recover spatial properties of scRNA-seq data by utilizing spatial measurements of a relatively small number of genes. A spatial metric for individual cells in scRNA-seq data is first established based on a map connecting it with the spatial measurements. The cell-cell communications are then obtained by "optimally transporting" signal senders to target signal receivers in space. Using partial information decomposition, we next compute the intercellular gene-gene information flow to estimate the spatial regulations between genes across cells. Four datasets are employed for cross-validation of spatial gene expression prediction and comparison to known cell-cell communications. SpaOTsc has broader applications, both in integrating non-spatial single-cell measurements with spatial data, and directly in spatial single-cell transcriptomics data to reconstruct spatial cellular dynamics in tissues.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
