

When genetic burden reaches threshold
- PMID: 32350504
- PMCID: PMC7599032
- DOI: 10.1093/eurheartj/ehaa269
When genetic burden reaches threshold
Abstract
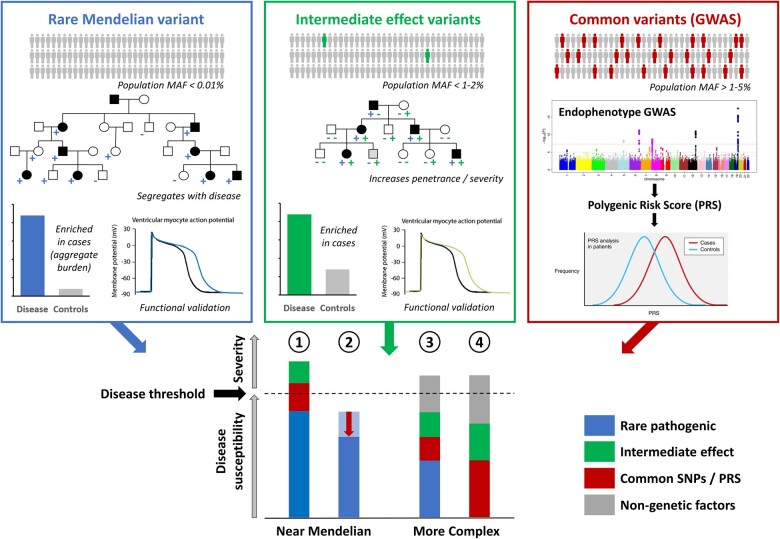
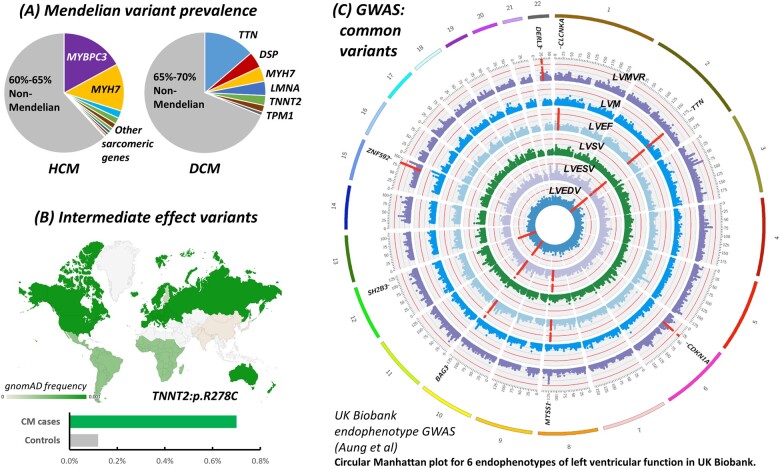
Rare cardiac genetic diseases have generally been considered to be broadly Mendelian in nature, with clinical genetic testing for these conditions predicated on the detection of a primary causative rare pathogenic variant that will enable cascade genetic screening in families. However, substantial variability in penetrance and disease severity among carriers of pathogenic variants, as well as the inability to detect rare Mendelian variants in considerable proportions of patients, indicates that more complex aetiologies are likely to underlie these diseases. Recent findings have suggested genetic variants across a range of population frequencies and effect sizes may combine, along with non-genetic factors, to determine whether the threshold for expression of disease is reached and the severity of the phenotype. The availability of increasingly large genetically characterized cohorts of patients with rare cardiac diseases is enabling the discovery of common genetic variation that may underlie both variable penetrance in Mendelian diseases and the genetic aetiology of apparently non-Mendelian rare cardiac conditions. It is likely that the genetic architecture of rare cardiac diseases will vary considerably between different conditions as well as between patients with similar phenotypes, ranging from near-Mendelian disease to models more akin to common, complex disease. Uncovering the broad range of genetic factors that predispose patients to rare cardiac diseases offers the promise of improved risk prediction and more focused clinical management in patients and their families.
Keywords: Genetic modifiers; Genetics; Genome-wide association studies; Inherited cardiomyopathies; Rare cardiac disease; Ventricular arrhythmias.
© The Author(s) 2020. Published by Oxford University Press on behalf of the European Society of Cardiology.
Figures
References
-
- Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJR, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–628. - PMC - PubMed
-
- Wordsworth S, Leal J, Blair E, Legood R, Thomson K, Seller A, Taylor J, Watkins H. DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J 2010;31:926–935. - PubMed
-
- Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res 2015;116:1919–1936. - PubMed
-
- Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, Jamal SM, Szybowska M, Morel CF, Bowdin S, Garcia J, Care M, Sturm AC, Novelli V, Ackerman MJ, Ware JS, Hershberger RE, Wilde AAM, Gollob MH; National Institutes of Health Clinical Genome Resource Consortium. Reappraisal of reported genes for sudden arrhythmic death. Circulation 2018;138:1195–1205. - PMC - PubMed
-
- Walsh R, Buchan R, Wilk A, John S, Felkin LE, Thomson KL, Chiaw TH, Loong CCW, Pua CJ, Raphael C, Prasad S, Barton PJ, Funke B, Watkins H, Ware JS, Cook SA. Defining the genetic architecture of hypertrophic cardiomyopathy: re-evaluating the role of non-sarcomeric genes. Eur Heart J 2017;38:3461–3468. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
