

Genetic predispositions of Parkinson's disease revealed in patient-derived brain cells
- PMID: 32352027
- PMCID: PMC7181694
- DOI: 10.1038/s41531-020-0110-8
Genetic predispositions of Parkinson's disease revealed in patient-derived brain cells
Abstract
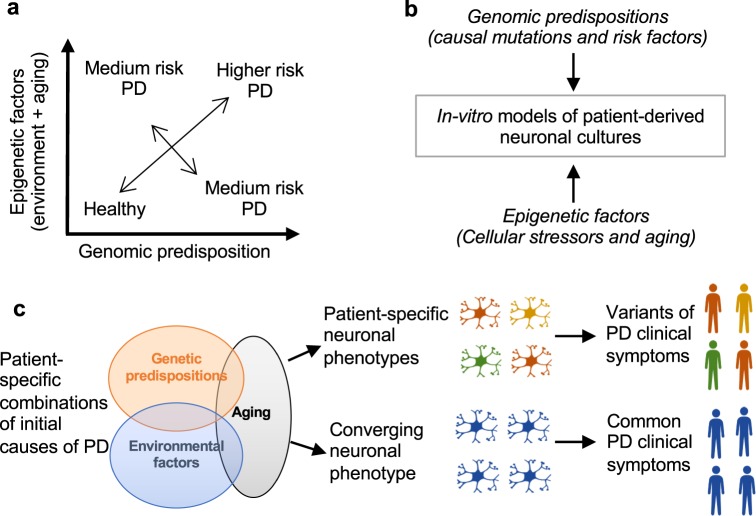
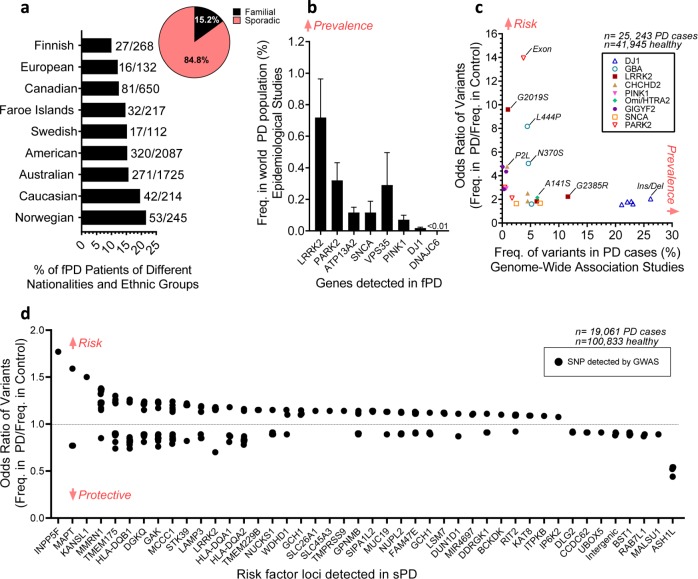
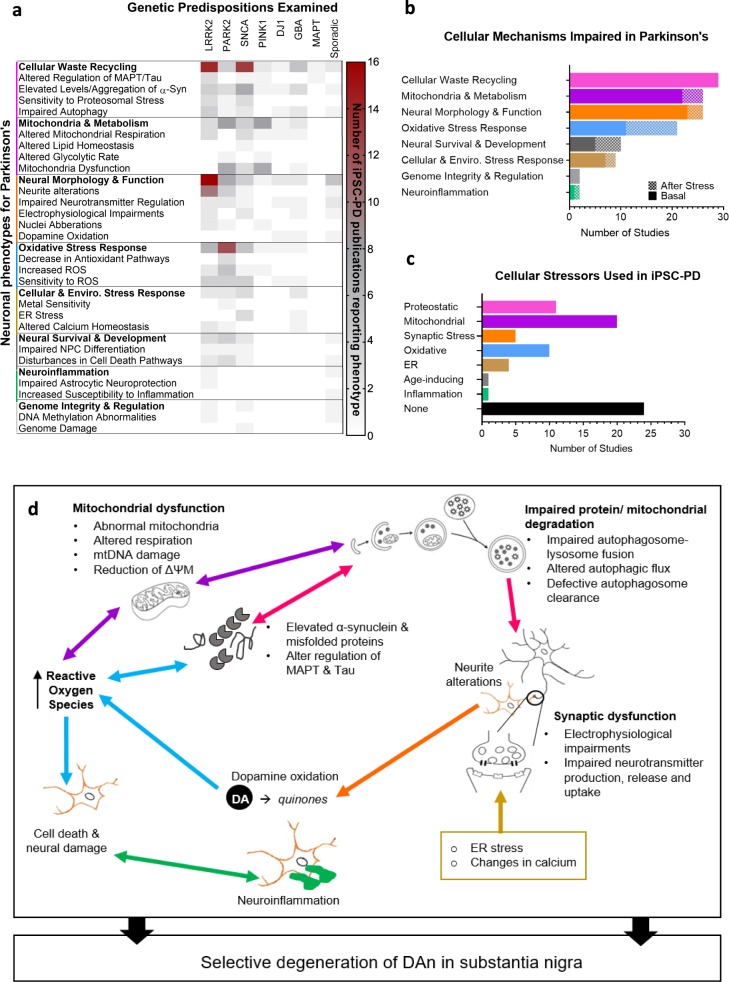
Parkinson's disease (PD) is the second most prevalent neurological disorder and has been the focus of intense investigations to understand its etiology and progression, but it still lacks a cure. Modeling diseases of the central nervous system in vitro with human induced pluripotent stem cells (hiPSC) is still in its infancy but has the potential to expedite the discovery and validation of new treatments. Here, we discuss the interplay between genetic predispositions and midbrain neuronal impairments in people living with PD. We first summarize the prevalence of causal Parkinson's genes and risk factors reported in 74 epidemiological and genomic studies. We then present a meta-analysis of 385 hiPSC-derived neuronal lines from 67 recent independent original research articles, which point towards specific impairments in neurons from Parkinson's patients, within the context of genetic predispositions. Despite the heterogeneous nature of the disease, current iPSC models reveal converging molecular pathways underlying neurodegeneration in a range of familial and sporadic forms of Parkinson's disease. Altogether, consolidating our understanding of robust cellular phenotypes across genetic cohorts of Parkinson's patients may guide future personalized drug screens in preclinical research.
Keywords: Cellular neuroscience; Induced pluripotent stem cells.
© The Author(s) 2020.
Conflict of interest statement
Competing interestsThe authors declare no competing interests.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources
