

Catalytic N2-to-NH3 (or -N2H4) Conversion by Well-Defined Molecular Coordination Complexes
- PMID: 32352271
- PMCID: PMC7493999
- DOI: 10.1021/acs.chemrev.9b00638
Catalytic N2-to-NH3 (or -N2H4) Conversion by Well-Defined Molecular Coordination Complexes
Abstract
Nitrogen fixation, the six-electron/six-proton reduction of N2, to give NH3, is one of the most challenging and important chemical transformations. Notwithstanding the barriers associated with this reaction, significant progress has been made in developing molecular complexes that reduce N2 into its bioavailable form, NH3. This progress is driven by the dual aims of better understanding biological nitrogenases and improving upon industrial nitrogen fixation. In this review, we highlight both mechanistic understanding of nitrogen fixation that has been developed, as well as advances in yields, efficiencies, and rates that make molecular alternatives to nitrogen fixation increasingly appealing. We begin with a historical discussion of N2 functionalization chemistry that traverses a timeline of events leading up to the discovery of the first bona fide molecular catalyst system and follow with a comprehensive overview of d-block compounds that have been targeted as catalysts up to and including 2019. We end with a summary of lessons learned from this significant research effort and last offer a discussion of key remaining challenges in the field.
Conflict of interest statement
The authors declare no competing financial interest.
Figures













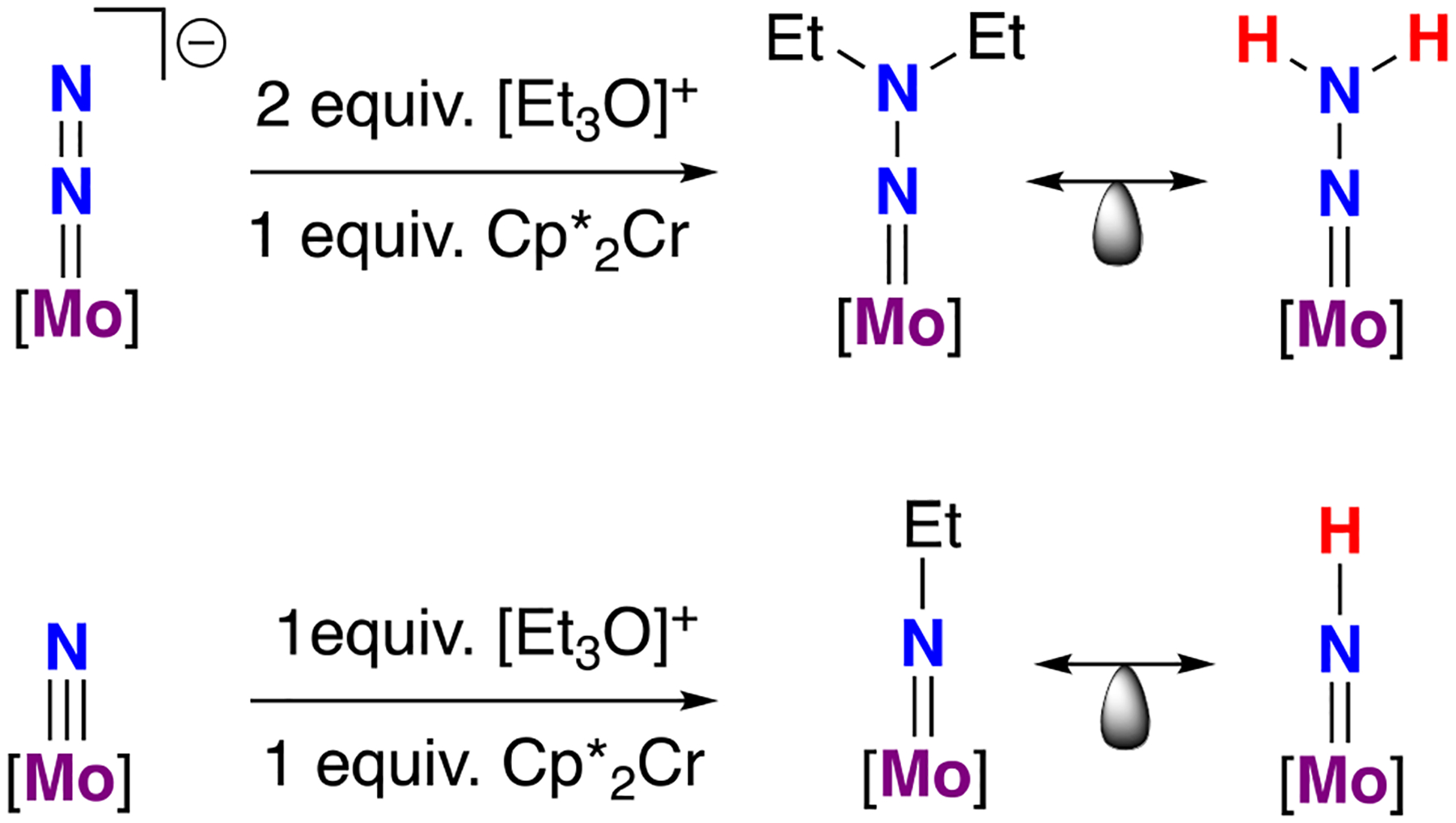


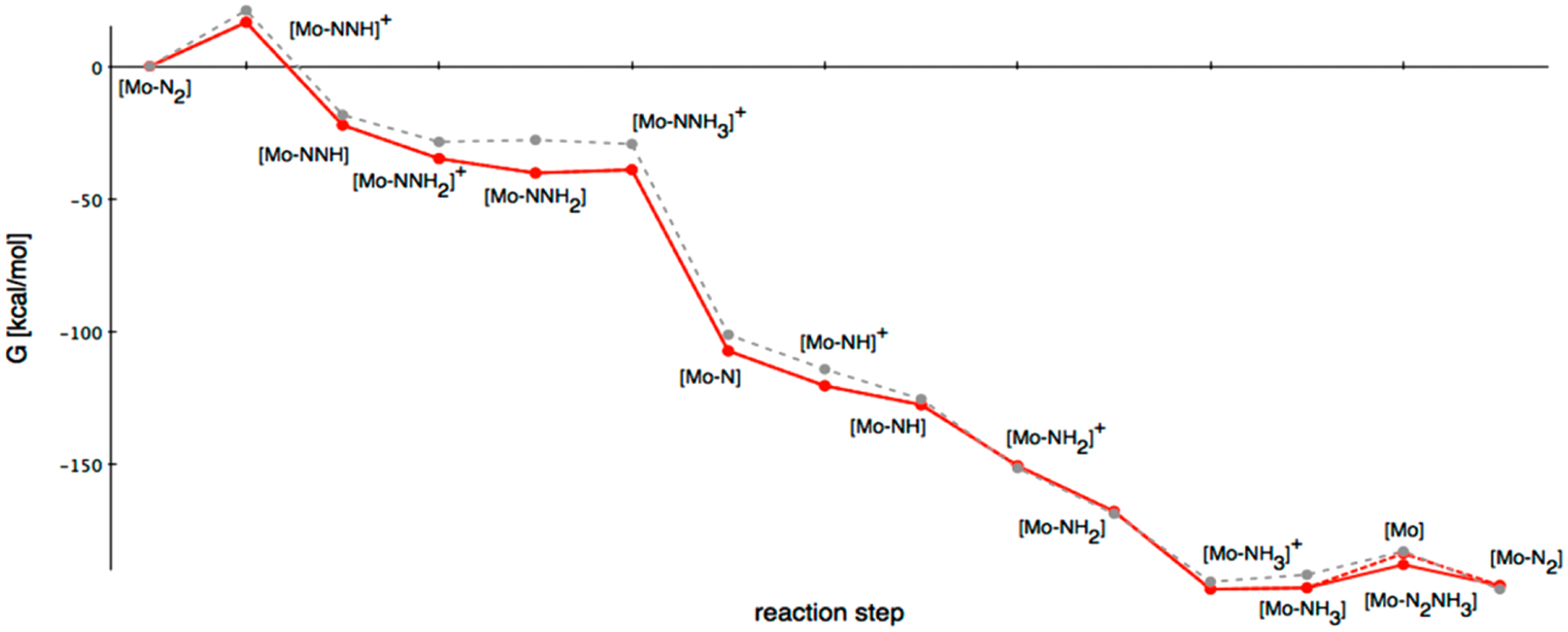


























































References
-
- Smil V Global Population and the Nitrogen Cycle. Sci. Am 1997, 277, 76–81.
-
- Schlögl R Catalytic Synthesis of Ammonia—A “Never-Ending Story”? Angew. Chem., Int. Ed 2003, 42, 2004–2008. - PubMed
-
- Appl M Ammonia http://www.iipinetwork.org/wp-content/Ietd/content/ammonia.html#technolo... (accessed Jun 22, 2019).
-
- Mosier AR Environmental Challenges Associated with Needed Increases in Global Nitrogen Fixation. Nutr. Cycling Agroecosyst 2002, 63, 101–116.
-
- Canfield DE; Glazer AN; Falkowski PG The Evolution and Future of Earth’s Nitrogen Cycle. Science 2010, 330, 192–196. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
