

Combining signal and sequence to detect RNA polymerase initiation in ATAC-seq data
- PMID: 32353042
- PMCID: PMC7192442
- DOI: 10.1371/journal.pone.0232332
Combining signal and sequence to detect RNA polymerase initiation in ATAC-seq data
Abstract
The assay for transposase-accessible chromatin followed by sequencing (ATAC-seq) is an inexpensive protocol for measuring open chromatin regions. ATAC-seq is also relatively simple and requires fewer cells than many other high-throughput sequencing protocols. Therefore, it is tractable in numerous settings where other high throughput assays are challenging to impossible. Hence it is important to understand the limits of what can be inferred from ATAC-seq data. In this work, we leverage ATAC-seq to predict the presence of nascent transcription. Nascent transcription assays are the current gold standard for identifying regions of active transcription, including markers for functional transcription factor (TF) binding. We combine mapped short reads from ATAC-seq with the underlying peak sequence, to determine regions of active transcription genome-wide. We show that a hybrid signal/sequence representation classified using recurrent neural networks (RNNs) can identify these regions across different cell types.
Conflict of interest statement
One author (RDD) of this publication is a founder and scientific advisor for Arpeggio Biosciences. Dr. Dowell is not employed by Arpeggio but rather consults occasionally with the company. We also note that no aspect of this work was funded by or influenced in any way by the company. This work is funded entirely by NIH R01 GM125871. No aspect of our funding alters our adherence to PLOS ONE policies on sharing data and materials.
Figures

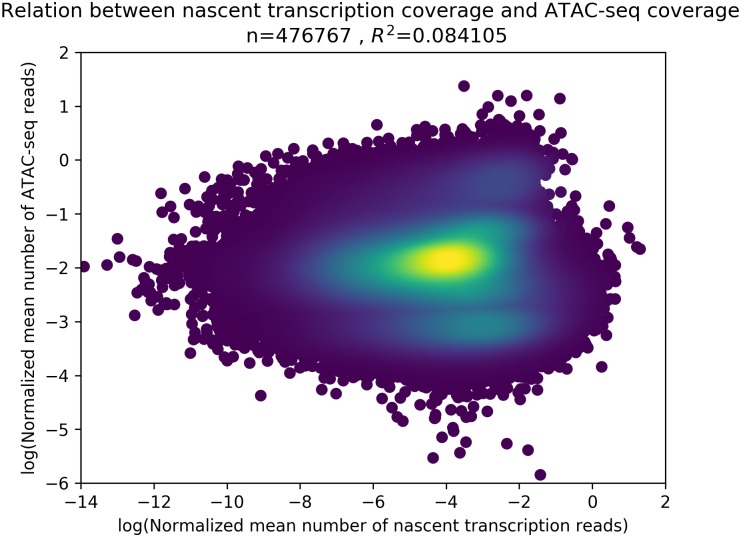




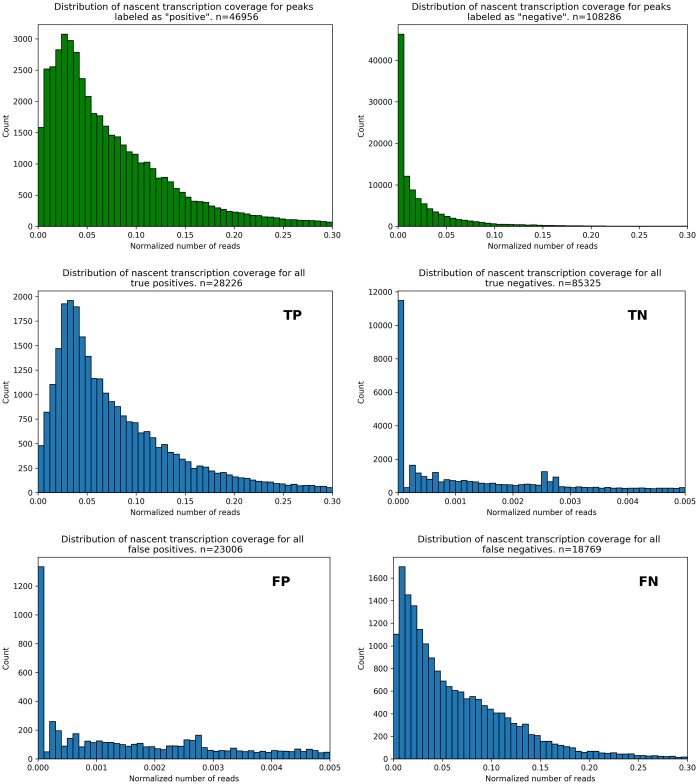
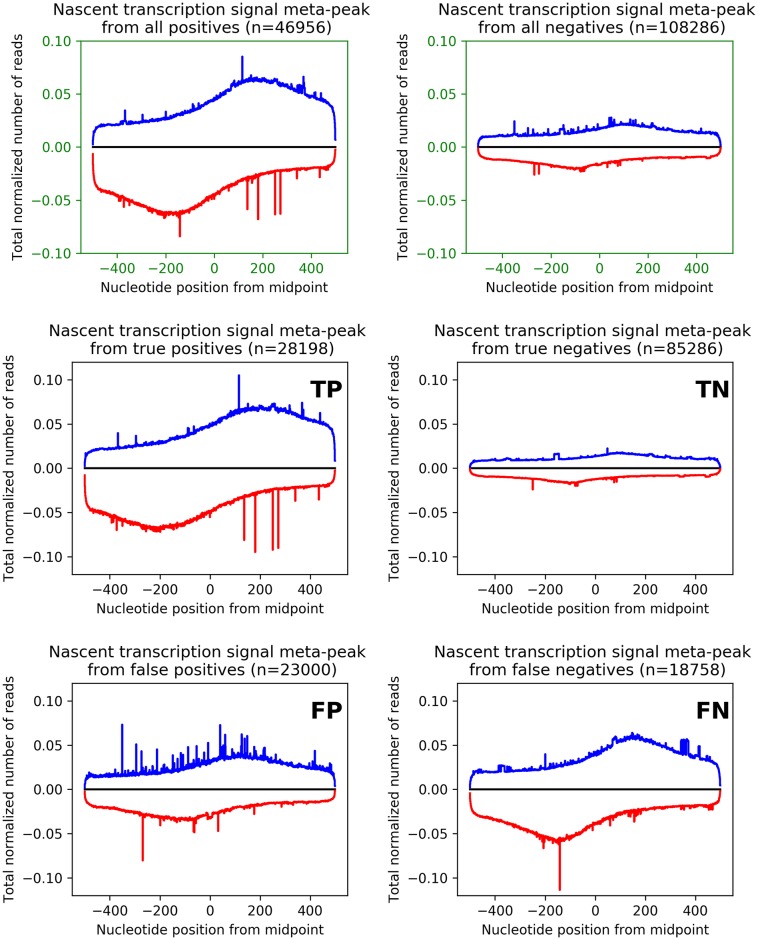


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
