

Two novel ECHS1 variants, affecting splicing and reducing enzyme activity, is associated with mitochondrial encephalopathy in infant: a case report
- PMID: 32354323
- PMCID: PMC7193542
- DOI: 10.1186/s12883-020-01735-y
Two novel ECHS1 variants, affecting splicing and reducing enzyme activity, is associated with mitochondrial encephalopathy in infant: a case report
Abstract
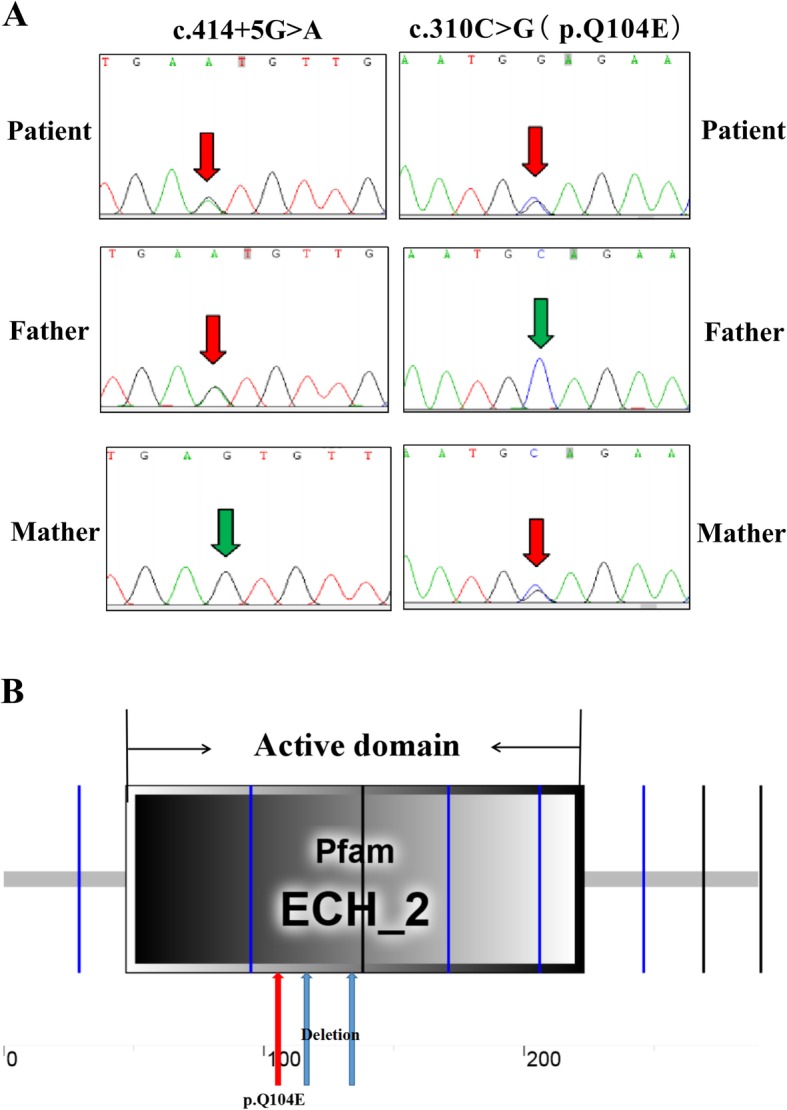
Background: Short-chain enoyl-CoA hydratase (ECHS1) is a multifunctional mitochondrial matrix enzyme involved in the second step of mitochondrial fatty acid β-oxidation. Mitochondrial diseases resulting from ECHS1 mutations are often characterised by encephalopathy, deafness, epilepsy, optic atrophy, cardiomyopathy, dystonia, and lactic acidosis. In this study, we report two novel heterogeneous variants, c.414 + 5G > A (in intron 3) and c.310C > G (in CDS), of ECHS1 in an infant with mitochondrial encephalopathy.
Case presentation: The two novel variants, c.414 + 5G > A (Chr10:135183403) in intron 3 and c.310C > G (Chr10:135183512) in CDS, were identified by next generation sequencing (NGS). A minigene assay was used to analyse the function of the c.414 + 5G > A variant. ECHS1 enzyme activity was measured by spectrophotometry in the patient-derived myoblasts. The 2-year old patient presented with mitochondrial encephalopathy since birth. Clinical features were encephalopathy, epilepsy, and hindered psychomotor and language development. Serum lactate and blood ammonia levels were elevated, and brain magnetic resonance imaging showed abnormal signals in the bilateral frontal, parietal, and occipital cortices and brainstem and basal ganglia. We found two novel heterogeneous variants in ECHS1 in this patient. Minigene assay revealed the c.414 + 5G > A variant as the cause of intronic cryptic splice site activation and 39 bp deletion in mature mRNA. In silico analysis predicted that c.310C > G might change glutamine (Q) to glutamic acid (E) in the 104th amino acid sequence (p.Q104E). To investigate the impact of these two variants on protein function, we constructed a 3D model of human ECHS1 and showed that the variants might alter the highly conserved region in close proximity to the active site, which might hinder, or even halt, enzymatic activity. The experimental assay showed that ECHS1 enzyme activity in the patient-derived myoblasts decreased compared to that in control.
Conclusions: Our findings are the first to report a mitochondrial encephalopathy infant carrying two novel ECHS1 variants, c.414 + 5G > A and c.310C > G, which might be deleterious variants, function as pathogenicity markers for mitochondrial encephalopathy, and facilitate disease diagnosis.
Keywords: ECHS1; Mitochondrial encephalopathy; Variants.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
-
- Yamada K, Naiki M, Hoshino S, Kitaura Y, Kondo Y, Nomura N, Kimura R, Fukushi D, Yamada Y, Shimozawa N, Yamaguchi S, Shimomura Y, Miura K, Wakamatsu N. Clinical and biochemical characterization of 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) deficiency that causes Leigh-like disease and ketoacidosis. Mol Genet Metab Rep. 2014;1:455–460. doi: 10.1016/j.ymgmr.2014.10.003. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- 2016YFC1306202/The National Key Research and Development Program of China
- 2014-160/Wuhan Science and Technology Innovation Platform-Children Nerve Disease Clinical Medical Research Center Funded Projects
- FTCSF-2018-05/A Multicenter Clinical Study of Ketogenic Diet in the Treatment of Mitochondrial Epilepsy
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
