

The natural history of infantile neuroaxonal dystrophy
- PMID: 32357911
- PMCID: PMC7193406
- DOI: 10.1186/s13023-020-01355-2
The natural history of infantile neuroaxonal dystrophy
Abstract
Background: Infantile neuroaxonal dystrophy (INAD) is a rapidly progressive neurodegenerative disorder of early onset causing premature death. It results from biallelic pathogenic variants in PLA2G6, which encodes a calcium-independent phospholipase A2.
Objective: We aim to outline the natural history of INAD and provide a comprehensive description of its clinical, radiological, laboratory, and molecular findings.
Materials and methods: We comprehensively analyzed the charts of 28 patients: 16 patients from Riyadh, Saudi Arabia, 8 patients from North and South America and 4 patients from Europe with a molecularly confirmed diagnosis of PLA2G6-associated neurodegeneration (PLAN) and a clinical history consistent with INAD.
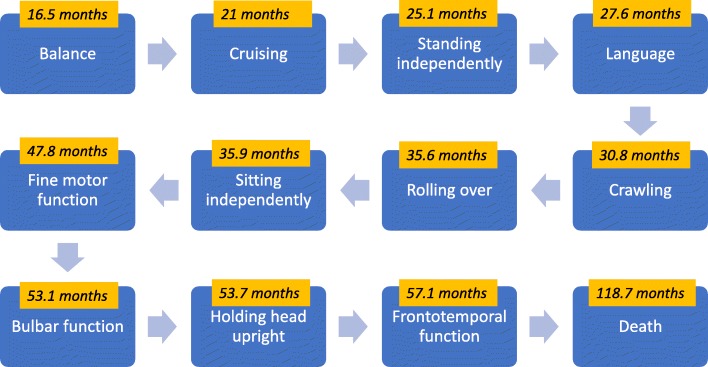
Results: In our cohort, speech impairment and loss of gross motor milestones were the earliest signs of the disease. As the disease progressed, loss of fine motor milestones and bulbar dysfunction were observed. Temporo-frontal function was among the last of the milestones to be lost. Appendicular spastic hypertonia, axial hypotonia, and hyperreflexia were common neurological findings. Other common clinical findings include nystagmus (60.7%), seizures (42.9%), gastrointestinal disease (42.9%), skeletal deformities (35.7%), and strabismus (28.6%). Cerebellar atrophy and elevations in serum AST and LDH levels were consistent features of INAD. There was a statistically significant difference when comparing patients with non-sense/truncating variants compared with missense/in-frame deletions in the time of initial concern (p = 0.04), initial loss of language (p = 0.001), initial loss of fine motor skills (p = 0.009), and initial loss of bulbar skills (p = 0.007).
Conclusion: INAD is an ultra-rare neurodegenerative disorder that presents in early childhood, with a relentlessly progressive clinical course. Knowledge of the natural history of INAD may serve as a resource for healthcare providers to develop a targeted care plan and may facilitate the design of clinical trials to treat this disease.
Keywords: INAD; Infantile neuroaxonal dystrophy; Molecular genetics; Natural history.
Conflict of interest statement
Peter Milner, Sarah Endemann, Mark Midei, Paldeep Atwal, are employed by, and hold stock in Retrotope, Inc. All other authors have no potential conflicts of interest. All authors confirm that the content of the article has not been influenced by the sponsor.
Figures


References
-
- Balsinde J, Balboa MAJCs. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A2 in activated cells. Cell Signal. 2005;17(9):1052–62. - PubMed
-
- Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk JJJoBC. Human pancreatic islets express mRNA species encoding two distinct catalytically active isoforms of group VI phospholipase A2 (iPLA2) that arise from an exon-skipping mechanism of alternative splicing of the transcript from the iPLA2 gene on chromosome 22q13. 1. J Biol Chem. 1999;274(14):9607–16. - PMC - PubMed
-
- Larsson Forsell PK, Kennedy BP, Claesson HEJEjob. The human calcium-independent phospholipase A2 gene: Multiple enzymes with distinct properties from a single gene. Eur J Biochem. 1999;262(2):575–85. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
