

Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China
- PMID: 32359424
- PMCID: PMC7192124
- DOI: 10.1016/j.cell.2020.04.023
Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China
Abstract
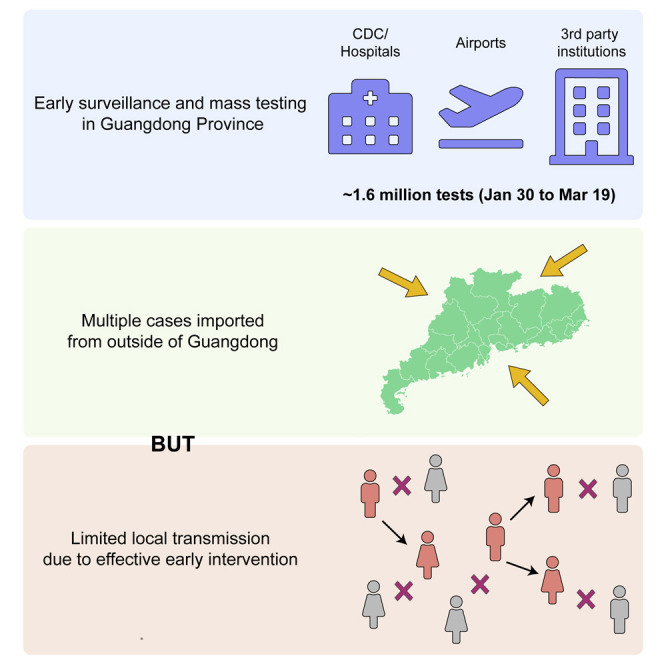
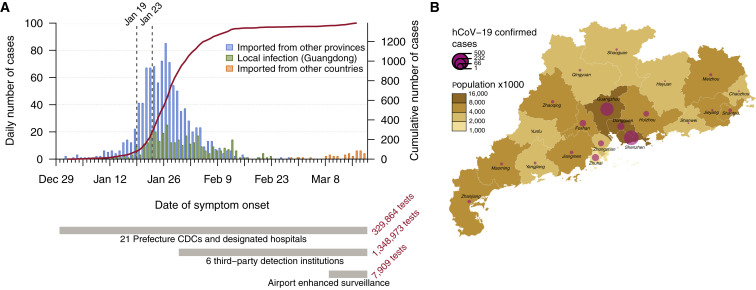
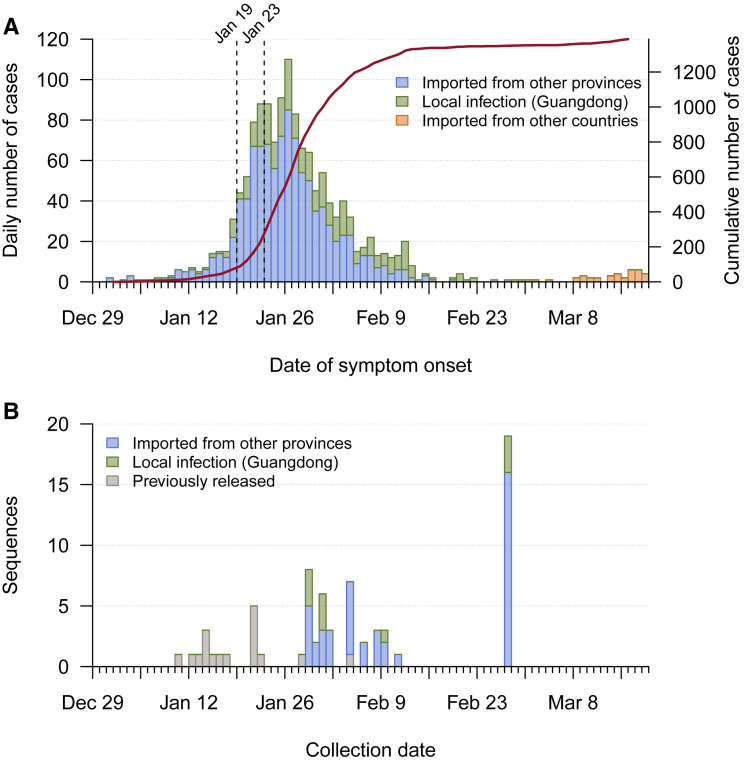
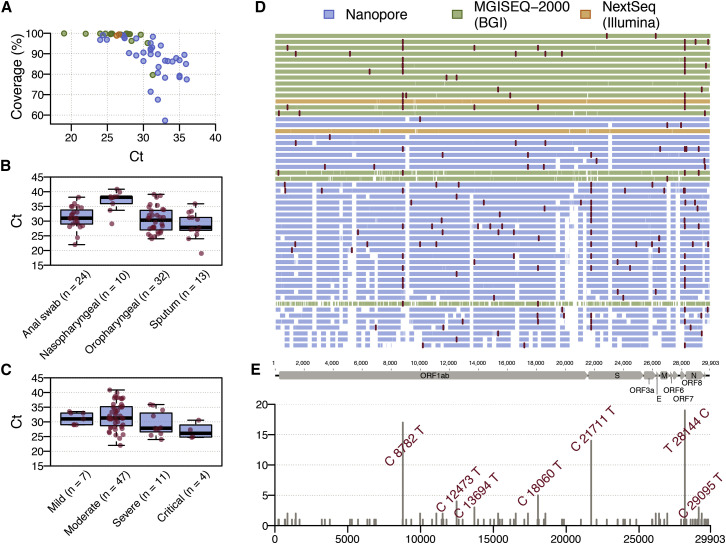
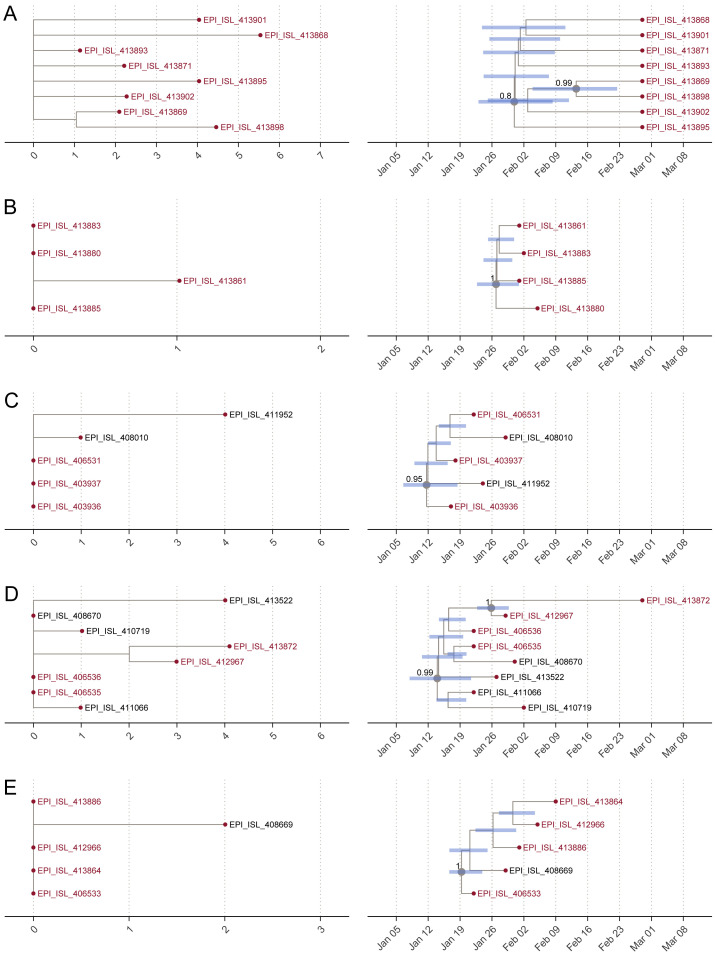
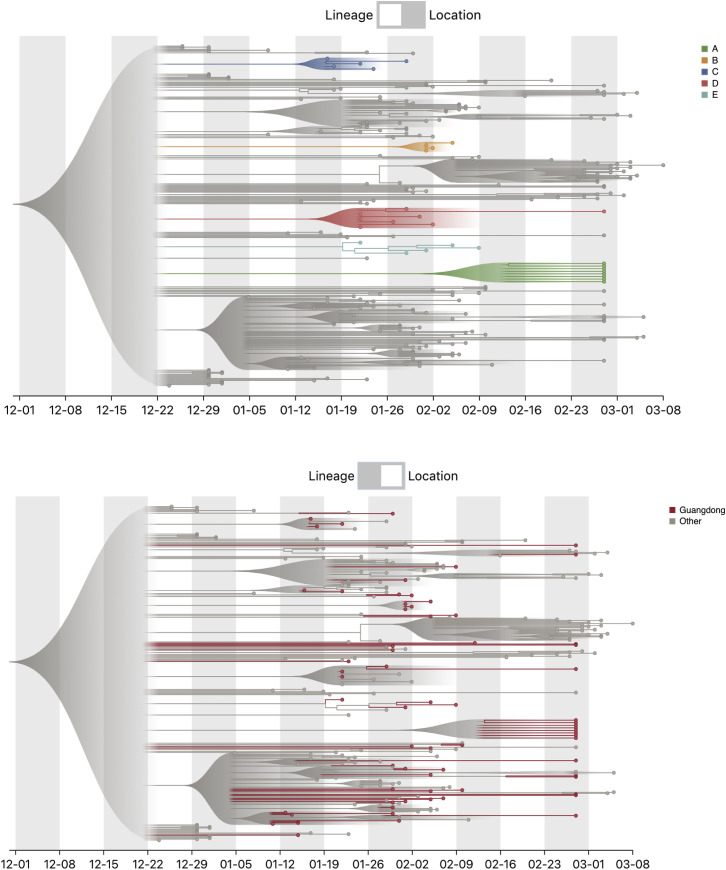
Coronavirus disease 2019 (COVID-19) is caused by SARS-CoV-2 infection and was first reported in central China in December 2019. Extensive molecular surveillance in Guangdong, China's most populous province, during early 2020 resulted in 1,388 reported RNA-positive cases from 1.6 million tests. In order to understand the molecular epidemiology and genetic diversity of SARS-CoV-2 in China, we generated 53 genomes from infected individuals in Guangdong using a combination of metagenomic sequencing and tiling amplicon approaches. Combined epidemiological and phylogenetic analyses indicate multiple independent introductions to Guangdong, although phylogenetic clustering is uncertain because of low virus genetic variation early in the pandemic. Our results illustrate how the timing, size, and duration of putative local transmission chains were constrained by national travel restrictions and by the province's large-scale intensive surveillance and intervention measures. Despite these successes, COVID-19 surveillance in Guangdong is still required, because the number of cases imported from other countries has increased.
Keywords: COVID-19; SARS-COV-2; genomic epidemiology; phylogenetics; real-time disease surveillance; virus evolution.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
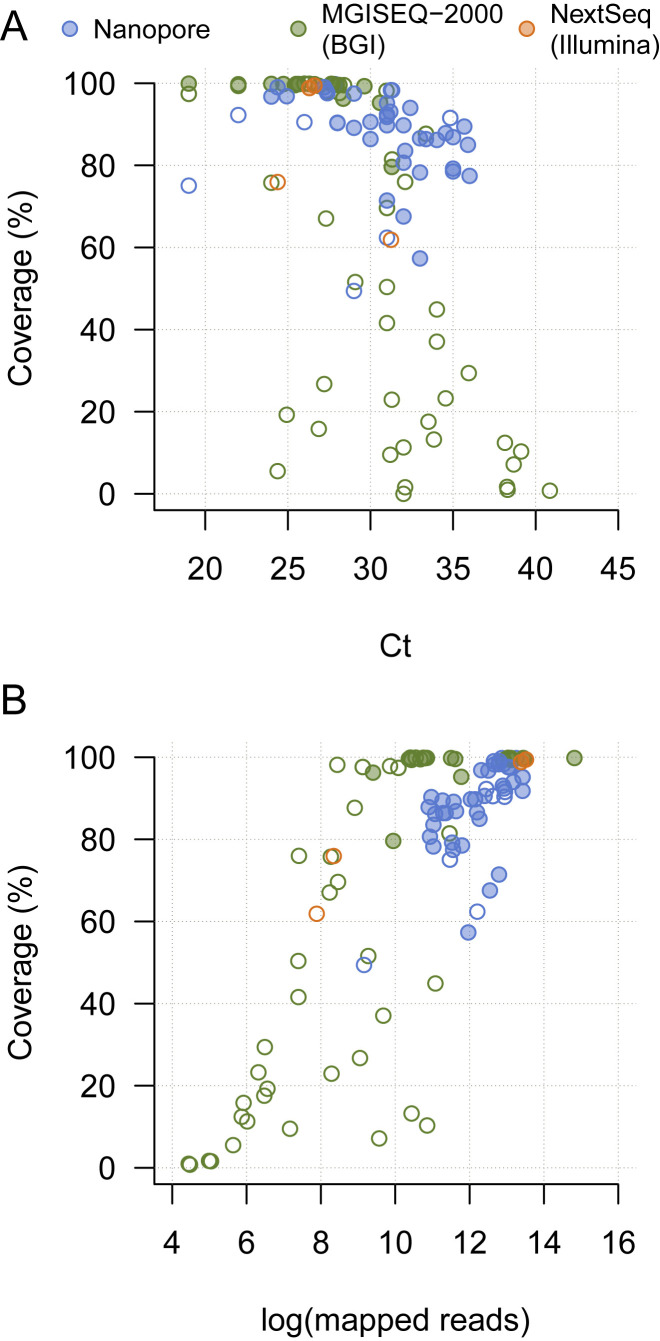
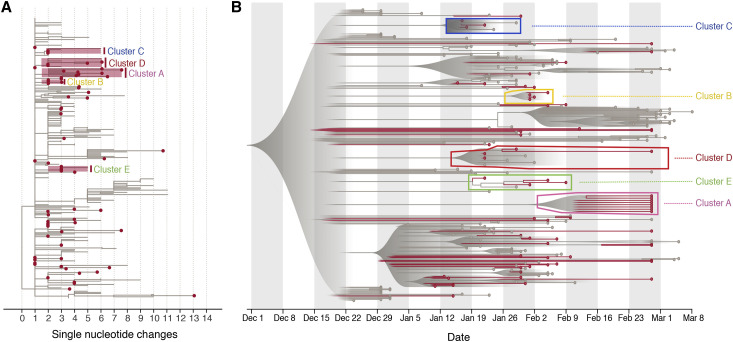
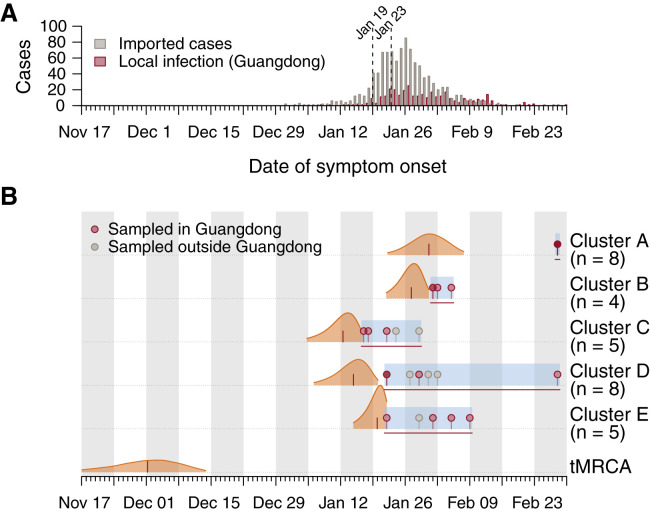
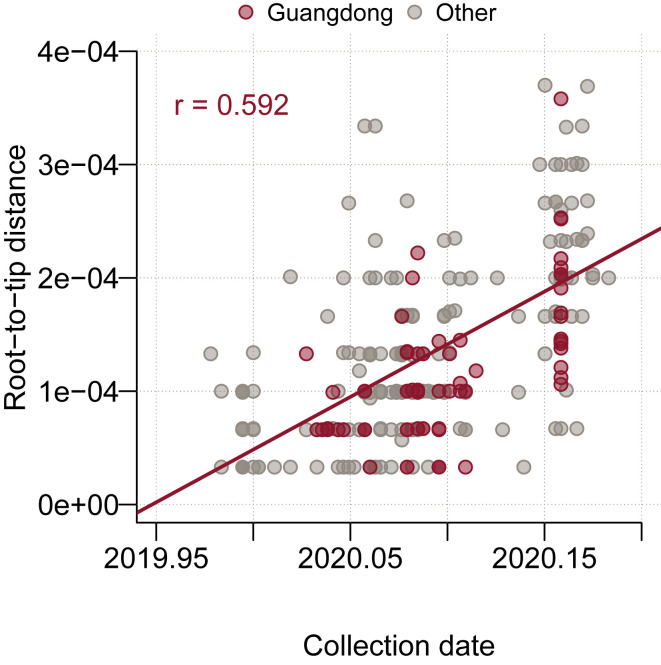
Comment in
-
Tracking the Genomic Footprints of SARS-CoV-2 Transmission.Trends Genet. 2020 Aug;36(8):544-546. doi: 10.1016/j.tig.2020.05.009. Epub 2020 May 28. Trends Genet. 2020. PMID: 32527617 Free PMC article.
References
-
- Ferreira M.A., Suchard M.A. Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 2008;36:355–368.
-
- Flightradar24 . 2020. Air traffic at China’s busiest airports down 80% since the beginning of the year.https://www.flightradar24.com/blog/air-traffic-at-chinas-busiest-airport...
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
