

Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis
- PMID: 32362324
- PMCID: PMC7305990
- DOI: 10.1016/j.immuni.2020.04.001
Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis
Abstract
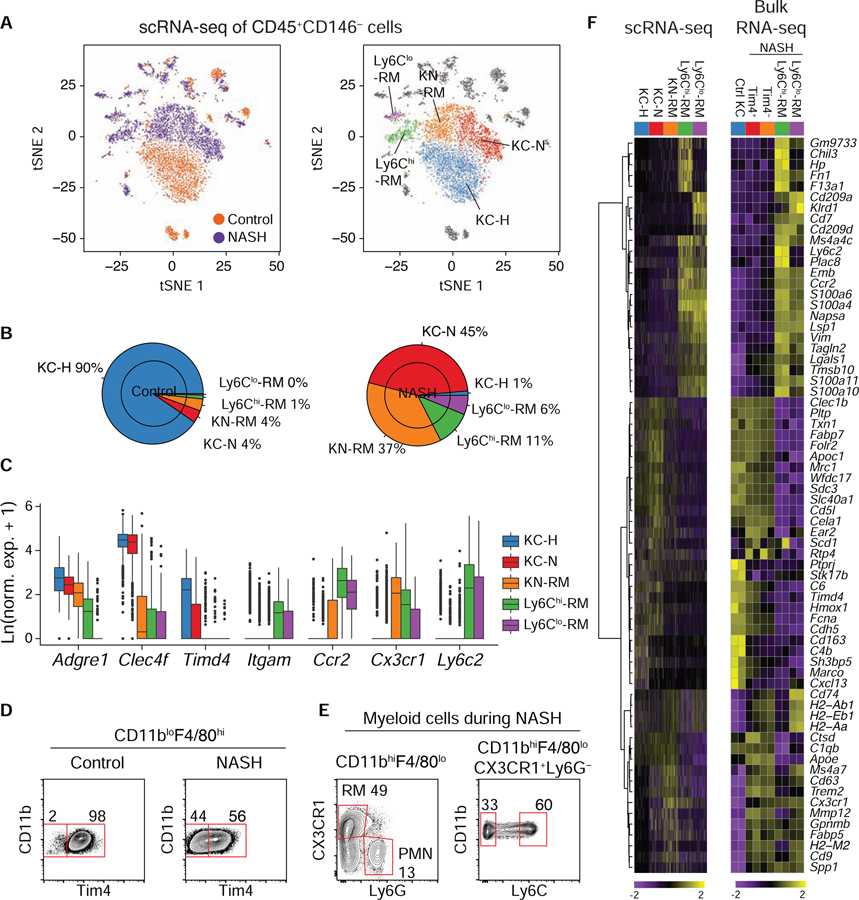
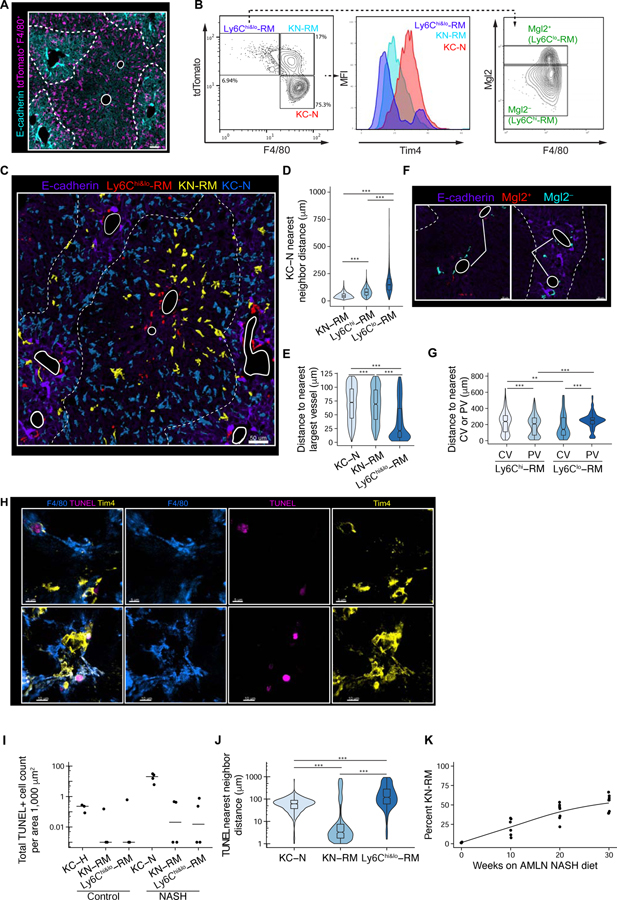
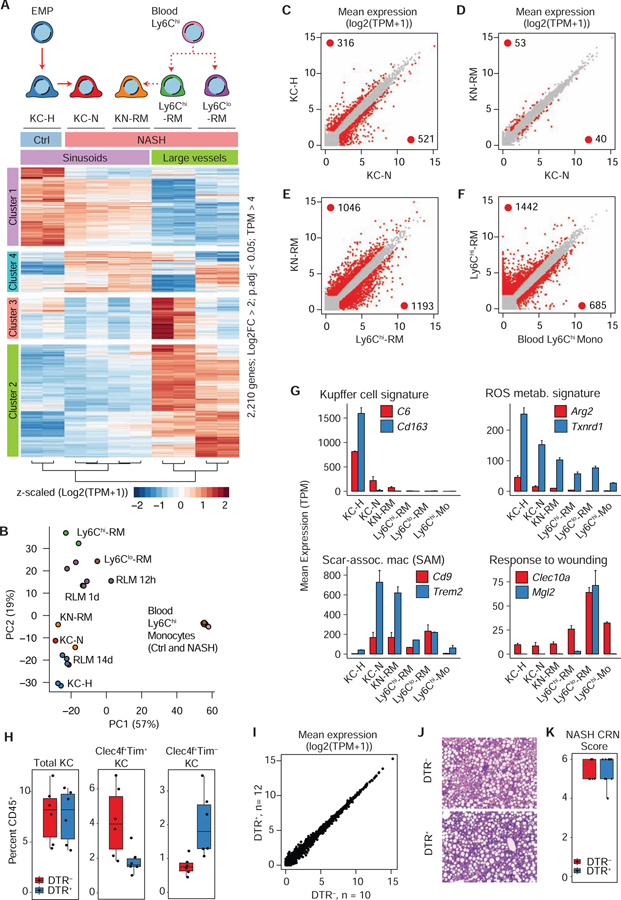
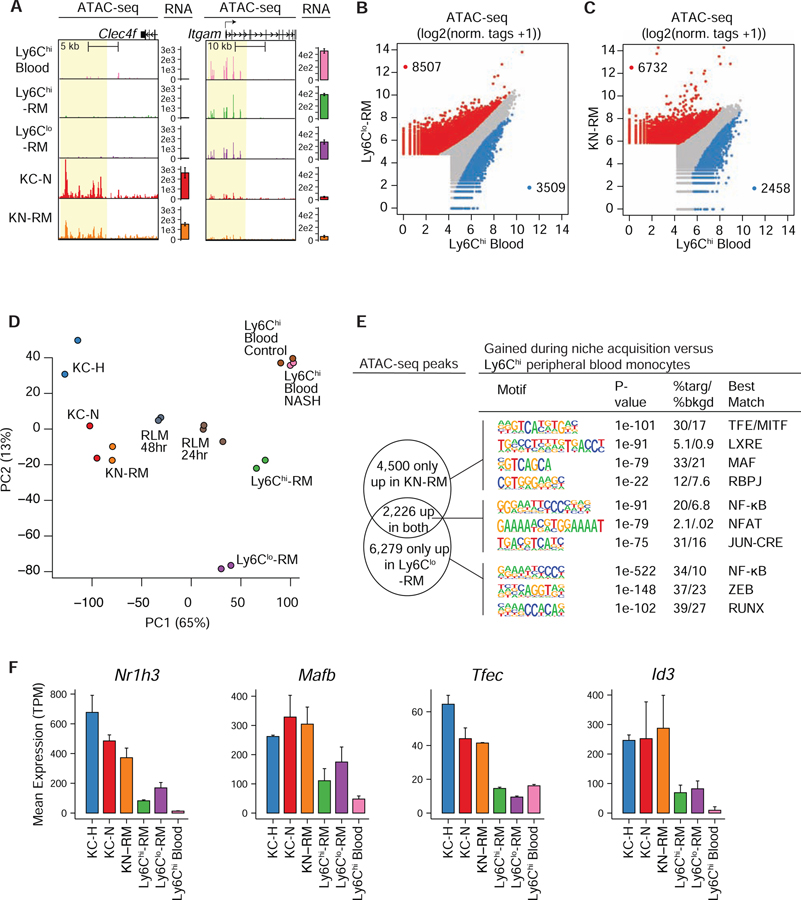
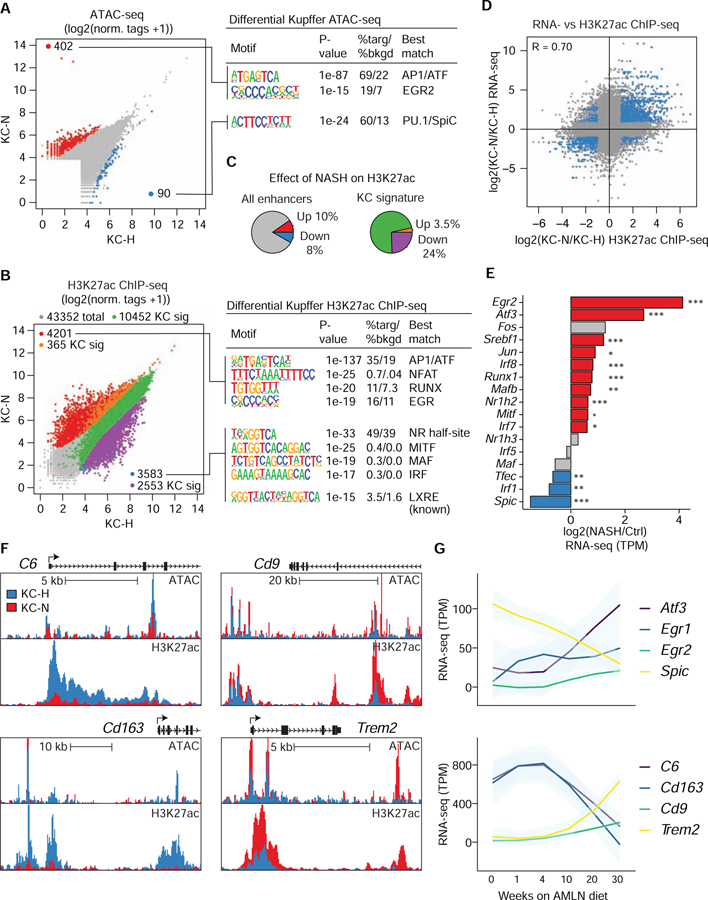
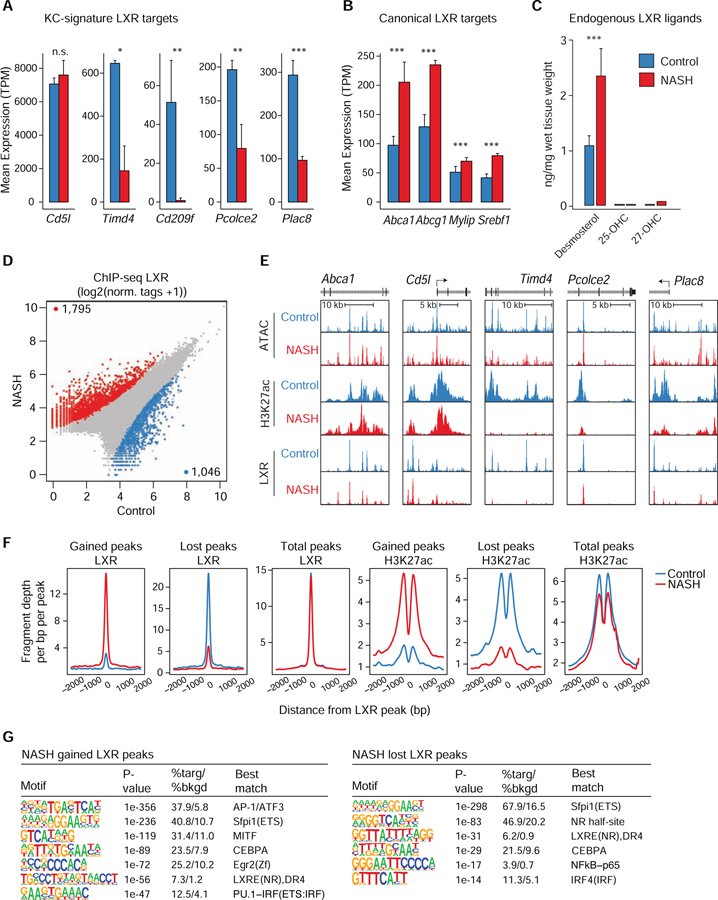
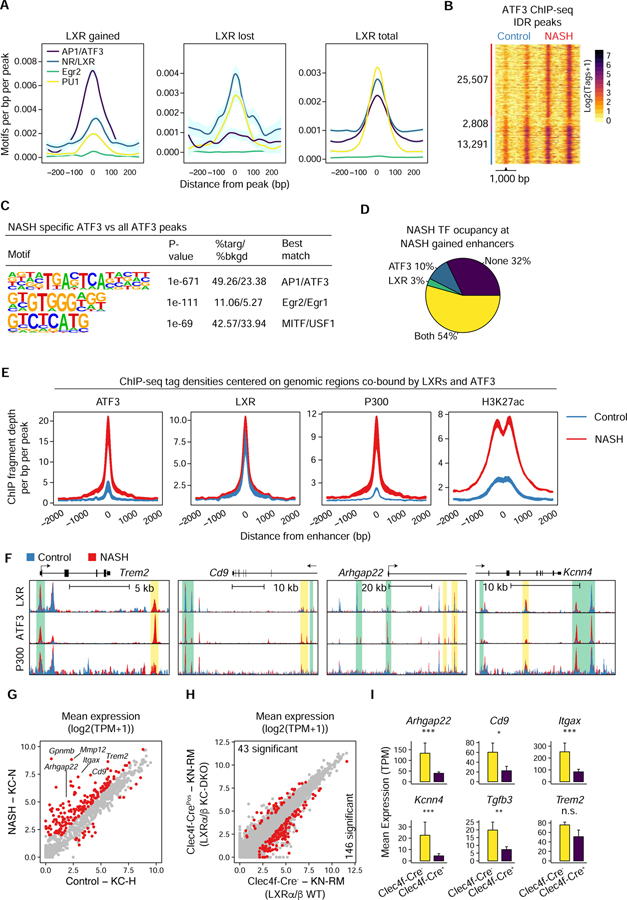
Tissue-resident and recruited macrophages contribute to both host defense and pathology. Multiple macrophage phenotypes are represented in diseased tissues, but we lack deep understanding of mechanisms controlling diversification. Here, we investigate origins and epigenetic trajectories of hepatic macrophages during diet-induced non-alcoholic steatohepatitis (NASH). The NASH diet induced significant changes in Kupffer cell enhancers and gene expression, resulting in partial loss of Kupffer cell identity, induction of Trem2 and Cd9 expression, and cell death. Kupffer cell loss was compensated by gain of adjacent monocyte-derived macrophages that exhibited convergent epigenomes, transcriptomes, and functions. NASH-induced changes in Kupffer cell enhancers were driven by AP-1 and EGR that reprogrammed LXR functions required for Kupffer cell identity and survival to instead drive a scar-associated macrophage phenotype. These findings reveal mechanisms by which disease-associated environmental signals instruct resident and recruited macrophages to acquire distinct gene expression programs and corresponding functions.
Keywords: ATF3; ChIP-seq; Kupffer cell; LXR; TREM2; epigenetics; genomics; nonalcoholic steatohepatitis; scRNA-seq; tissue macrophage.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests J.L.W. and X.S. are named inventors on patent applications or patents related to the use of oxidation-specific antibodies that are held by UCSD. J.L.W. is a consultant to Ionis Pharm and is a scientific founder of Oxitope, Inc.
Figures
Comment in
-
Not Chopped Liver-A Careful, Fate-Mapping Study of Macrophages in NASH.Cell Metab. 2020 Sep 1;32(3):328-330. doi: 10.1016/j.cmet.2020.08.005. Cell Metab. 2020. PMID: 32877688
References
-
- Baddeley A, Rubak E, and Turner R (2016). Spatial point patterns : methodology and applications with R (Boca Raton; London; New York: CRC Press, Taylor & Francis Group; ).
-
- Bonnardel J, T’Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, Vanneste B, De Prijck S, Nedospasov SA, Kremer A, et al. (2019). Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 51, 638–654 e639. - PMC - PubMed
-
- Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, and Waisman A (2005). A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods 2, 419–426. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 HL088093/HL/NHLBI NIH HHS/United States
- T32 DK007044/DK/NIDDK NIH HHS/United States
- R01 DK091183/DK/NIDDK NIH HHS/United States
- P30 DK063491/DK/NIDDK NIH HHS/United States
- T32 DK007202/DK/NIDDK NIH HHS/United States
- R01 GM065490/GM/NIGMS NIH HHS/United States
- P50 GM085764/GM/NIGMS NIH HHS/United States
- T32 CA009523/CA/NCI NIH HHS/United States
- K99 HL148504/HL/NHLBI NIH HHS/United States
- T32 DK007541/DK/NIDDK NIH HHS/United States
- R21 HL088083/HL/NHLBI NIH HHS/United States
- T32 GM007198/GM/NIGMS NIH HHS/United States
- P30 CA014195/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
