

Intracellular Trafficking Mechanisms of Synaptic Dysfunction in Alzheimer's Disease
- PMID: 32362813
- PMCID: PMC7180223
- DOI: 10.3389/fncel.2020.00072
Intracellular Trafficking Mechanisms of Synaptic Dysfunction in Alzheimer's Disease
Abstract
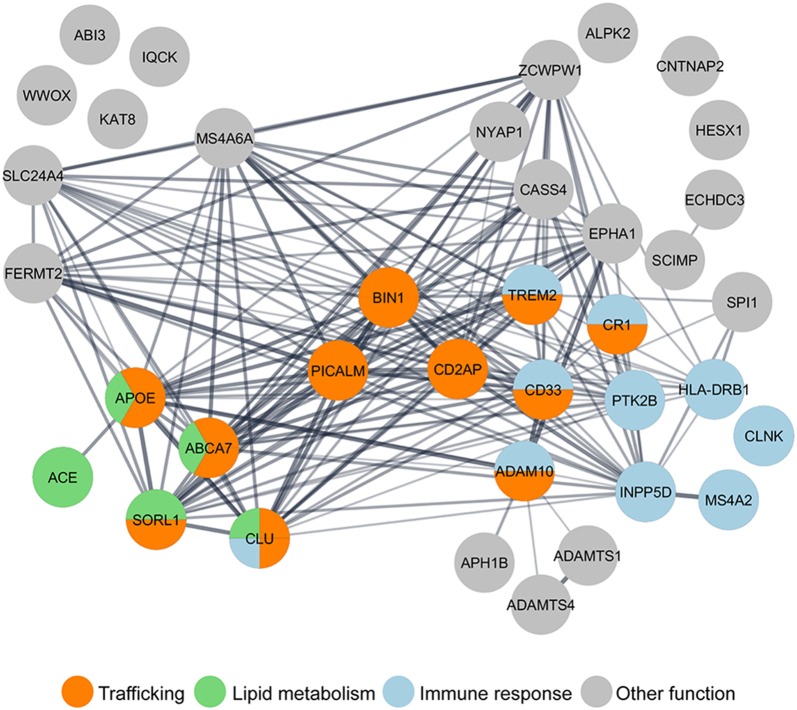
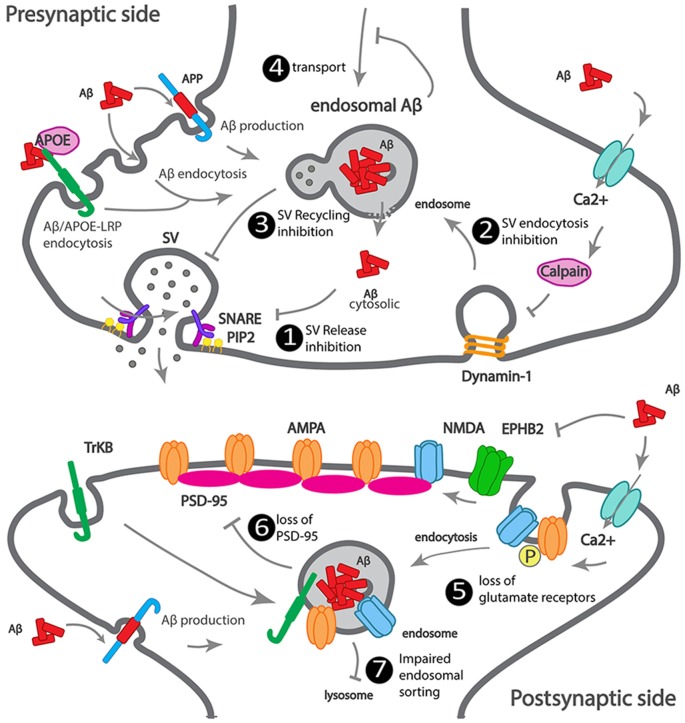
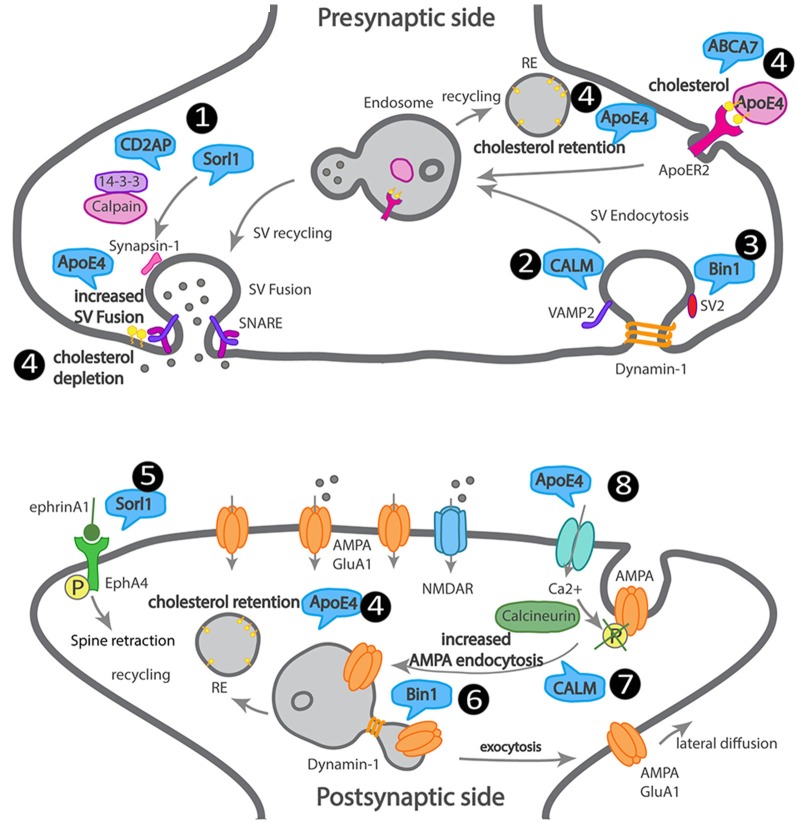
Alzheimer's disease (AD) is the most common neurodegenerative disease characterized by progressive memory loss. Although AD neuropathological hallmarks are extracellular amyloid plaques and intracellular tau tangles, the best correlate of disease progression is synapse loss. What causes synapse loss has been the focus of several researchers in the AD field. Synapses become dysfunctional before plaques and tangles form. Studies based on early-onset familial AD (eFAD) models have supported that synaptic transmission is depressed by β-amyloid (Aβ) triggered mechanisms. Since eFAD is rare, affecting only 1% of patients, research has shifted to the study of the most common late-onset AD (LOAD). Intracellular trafficking has emerged as one of the pathways of LOAD genes. Few studies have assessed the impact of trafficking LOAD genes on synapse dysfunction. Since endocytic traffic is essential for synaptic function, we reviewed Aβ-dependent and independent mechanisms of the earliest synaptic dysfunction in AD. We have focused on the role of intraneuronal and secreted Aβ oligomers, highlighting the dysfunction of endocytic trafficking as an Aβ-dependent mechanism of synapse dysfunction in AD. Here, we reviewed the LOAD trafficking genes APOE4, ABCA7, BIN1, CD2AP, PICALM, EPH1A, and SORL1, for which there is a synaptic link. We conclude that in eFAD and LOAD, the earliest synaptic dysfunctions are characterized by disruptions of the presynaptic vesicle exo- and endocytosis and of postsynaptic glutamate receptor endocytosis. While in eFAD synapse dysfunction seems to be triggered by Aβ, in LOAD, there might be a direct synaptic disruption by LOAD trafficking genes. To identify promising therapeutic targets and biomarkers of the earliest synaptic dysfunction in AD, it will be necessary to join efforts in further dissecting the mechanisms used by Aβ and by LOAD genes to disrupt synapses.
Keywords: APOE4; BIN1; CD2AP; PICALM; endocytosis; late-onset Alzheimer’s disease; synapses; β-amyloid.
Copyright © 2020 Perdigão, Barata, Araújo, Mirfakhar, Castanheira and Guimas Almeida.
Figures
Similar articles
-
Impact of late-onset Alzheimer's genetic risk factors on beta-amyloid endocytic production.Cell Mol Life Sci. 2018 Jul;75(14):2577-2589. doi: 10.1007/s00018-018-2825-9. Epub 2018 Apr 27. Cell Mol Life Sci. 2018. PMID: 29704008 Free PMC article. Review.
-
Amyloid Beta Oligomers Target to Extracellular and Intracellular Neuronal Synaptic Proteins in Alzheimer's Disease.Front Neurol. 2019 Nov 1;10:1140. doi: 10.3389/fneur.2019.01140. eCollection 2019. Front Neurol. 2019. PMID: 31736856 Free PMC article.
-
The synapse as a treatment avenue for Alzheimer's Disease.Mol Psychiatry. 2022 Jul;27(7):2940-2949. doi: 10.1038/s41380-022-01565-z. Epub 2022 Apr 20. Mol Psychiatry. 2022. PMID: 35444256 Review.
-
Regulation of Synaptic Amyloid-β Generation through BACE1 Retrograde Transport in a Mouse Model of Alzheimer's Disease.J Neurosci. 2017 Mar 8;37(10):2639-2655. doi: 10.1523/JNEUROSCI.2851-16.2017. Epub 2017 Feb 3. J Neurosci. 2017. PMID: 28159908 Free PMC article.
-
Alzheimer's genetic risk factor Bin1 controls synapse vesicle exo-endocytosis in inhibitory synapses.Cell Rep. 2025 Jul 15;44(8):116005. doi: 10.1016/j.celrep.2025.116005. Online ahead of print. Cell Rep. 2025. PMID: 40674214
Cited by
-
Alzheimer's Disease: Tau Pathology and Dysfunction of Endocytosis.Front Mol Neurosci. 2021 Jan 22;13:583755. doi: 10.3389/fnmol.2020.583755. eCollection 2020. Front Mol Neurosci. 2021. PMID: 33551742 Free PMC article. No abstract available.
-
Synaptic tau: A pathological or physiological phenomenon?Acta Neuropathol Commun. 2021 Sep 9;9(1):149. doi: 10.1186/s40478-021-01246-y. Acta Neuropathol Commun. 2021. PMID: 34503576 Free PMC article. Review.
-
The Cleavage-Specific Tau 12A12mAb Exerts an Anti-Amyloidogenic Action by Modulating the Endocytic and Bioenergetic Pathways in Alzheimer's Disease Mouse Model.Int J Mol Sci. 2023 Jun 2;24(11):9683. doi: 10.3390/ijms24119683. Int J Mol Sci. 2023. PMID: 37298634 Free PMC article.
-
Organelle perturbation in Alzheimer's disease: do intracellular amyloid-ß and the fragmented Golgi mediate early intracellular neurotoxicity?Front Cell Dev Biol. 2025 Apr 15;13:1550211. doi: 10.3389/fcell.2025.1550211. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40302938 Free PMC article. Review.
-
Endocytosis and Alzheimer's disease.Geroscience. 2024 Feb;46(1):71-85. doi: 10.1007/s11357-023-00923-1. Epub 2023 Aug 30. Geroscience. 2024. PMID: 37646904 Free PMC article. Review.
References
-
- Alzheimer’s Association (2019). 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 15, 321–387. 10.1016/j.jalz.2019.01.010 - DOI
LinkOut - more resources
Full Text Sources
Miscellaneous