

Novel Compound Heterozygous Variants of ETHE1 Causing Ethylmalonic Encephalopathy in a Chinese Patient: A Case Report
- PMID: 32362910
- PMCID: PMC7181787
- DOI: 10.3389/fgene.2020.00341
Novel Compound Heterozygous Variants of ETHE1 Causing Ethylmalonic Encephalopathy in a Chinese Patient: A Case Report
Abstract
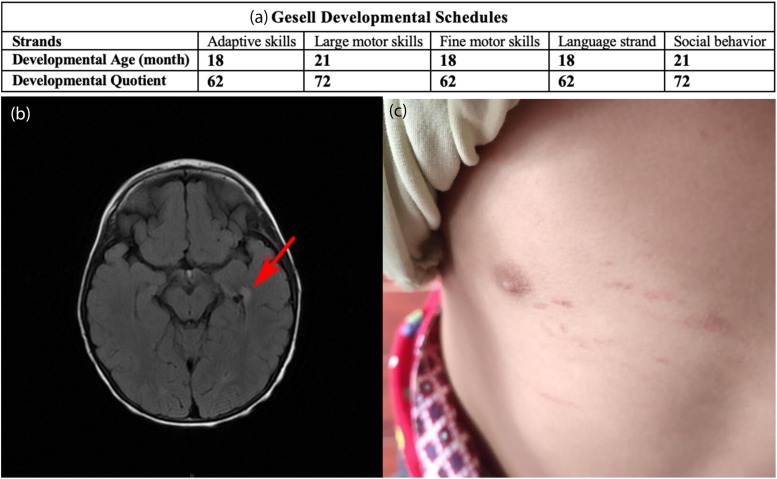
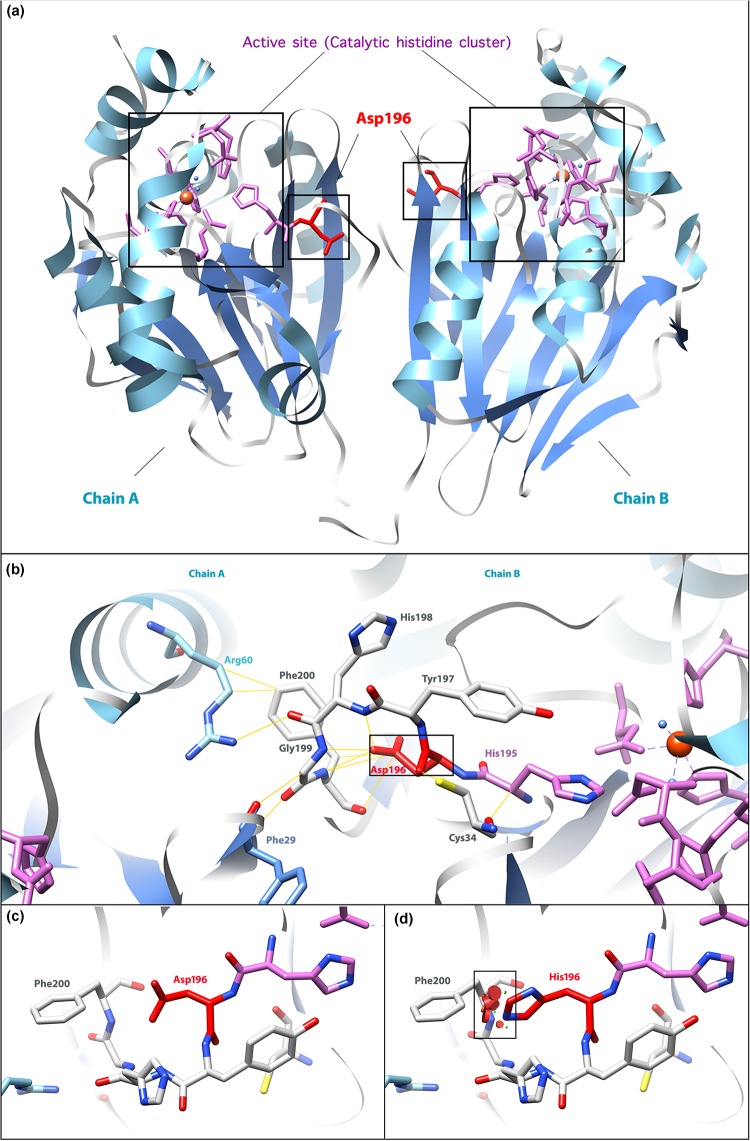
Ethylmalonic encephalopathy (EE) is a very rare autosomal recessive metabolic disorder that primarily affects children. Less than one hundred EE patients have been diagnosed worldwide. The clinical manifestations include chronic diarrhea, petechiae, orthostatic acrocyanosis, psychomotor delay and regression, seizures, and hypotonia. The ETHE1 gene has been shown to be associated with EE, and genetic sequencing provides concrete evidence for diagnosis. To date, only 37 variants of ETHE1 have been reported as disease-causing in EE patients. We identified two novel ETHE1 variants, i.e., c.595+1G>T at the canonical splice site and the missense variant c.586G>C (p. D196H), in a 3-year-old Chinese boy with EE. The patient had mild symptoms with only chronic diarrhea. The typical symptoms, including spontaneous petechiae, acrocyanosis, and hypotonia, were all absent. Herein, we report on the clinical, biochemical, and genetic findings of our patient and review the phenotypes and genotypes of all patients with EE caused by ETHE1 variants with available information. This study supports the early assessment and diagnosis of EE.
Keywords: ETHE1; chronic diarrhea; elevated ethylmalonic acid; ethylmalonic encephalopathy; genetic sequencing.
Copyright © 2020 Chen, Han and Yao.
Figures

Similar articles
-
Ethylmalonic encephalopathy: Clinical course and therapy response in an uncommon mild case with a severe ETHE1 mutation.Mol Genet Metab Rep. 2020 Aug 28;25:100641. doi: 10.1016/j.ymgmr.2020.100641. eCollection 2020 Dec. Mol Genet Metab Rep. 2020. PMID: 32923369 Free PMC article.
-
An atypically mild case of ethylmalonic encephalopathy with pathogenic ETHE1 variant.Am J Med Genet A. 2023 Jun;191(6):1614-1618. doi: 10.1002/ajmg.a.63176. Epub 2023 Mar 9. Am J Med Genet A. 2023. PMID: 36891747
-
Identification of a novel homozygous nonsense variant in a Chinese patient with ethylmalonic encephalopathy and a genotype-phenotype spectrum review.Clin Chim Acta. 2020 Oct;509:8-17. doi: 10.1016/j.cca.2020.05.051. Epub 2020 May 30. Clin Chim Acta. 2020. PMID: 32485156 Review.
-
ETHE1 mutations are specific to ethylmalonic encephalopathy.J Med Genet. 2006 Apr;43(4):340-6. doi: 10.1136/jmg.2005.036210. Epub 2005 Sep 23. J Med Genet. 2006. PMID: 16183799 Free PMC article.
-
Ethylmalonic Encephalopathy.2017 Sep 21. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2017 Sep 21. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 28933811 Free Books & Documents. Review.
Cited by
-
Case Report: Novel Compound-Heterozygous Variants of SKIV2L Gene that Cause Trichohepatoenteric Syndrome 2.Front Genet. 2021 Oct 6;12:756451. doi: 10.3389/fgene.2021.756451. eCollection 2021. Front Genet. 2021. PMID: 34691159 Free PMC article.
-
Red Flags in Primary Mitochondrial Diseases: What Should We Recognize?Int J Mol Sci. 2023 Nov 25;24(23):16746. doi: 10.3390/ijms242316746. Int J Mol Sci. 2023. PMID: 38069070 Free PMC article. Review.
-
Ethylmalonic encephalopathy: Clinical course and therapy response in an uncommon mild case with a severe ETHE1 mutation.Mol Genet Metab Rep. 2020 Aug 28;25:100641. doi: 10.1016/j.ymgmr.2020.100641. eCollection 2020 Dec. Mol Genet Metab Rep. 2020. PMID: 32923369 Free PMC article.
References
-
- Bijarnia-Mahay S., Gupta D., Shigematsu Y., Yamaguchi S., Saxena R., Verma I. C. (2016). Ethylmalonic encephalopathy in an Indian boy. Indian Pediatr. 53 914–916. - PubMed
-
- Burlina A., Zacchello F., Dionisi-Vici C., Bertini E., Sabetta G., Bennet M. J., et al. (1991). New clinical phenotype of branched-chain acyl-CoA oxidation defect. Lancet 338 1522–1523. - PubMed
-
- Choi Y. (2012). “A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein,” in Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, (Orlando, FL: ACM; ).
LinkOut - more resources
Full Text Sources