

Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2
- PMID: 32364041
- PMCID: PMC7232881
- DOI: 10.1080/07391102.2020.1764392
Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2
Abstract
SARS-CoV-2 is a new generation of coronavirus, which was first determined in Wuhan, China, in December 2019. So far, however, there no effective treatment has been found to stop this new generation of coronavirus but discovering of the crystal structure of SARS-CoV-2 main protease (SARS-CoV-2 Mpro) may facilitate searching for new therapies for SARS-COV-2. The aim was to assess the effectiveness of available FDA approved drugs which can construct a covalent bond with Cys145 inside binding site SARS-CoV-2 main protease by using covalent docking screening. We conducted the covdock module MMGBSA module in the Schrodinger suite 2020-1, to examine the covalent bonding utilizing. Besides, we submitted the top three drugs to molecular dynamics simulations via Gromacs 2018.1. The covalent docking showed that saquinavir, ritonavir, remdesivir, delavirdine, cefuroxime axetil, oseltamivir and prevacid have the highest binding energies MMGBSA of -72.17, -72.02, -65.19, -57.65, -54.25, -51.8, and -51.14 kcal/mol, respectively. The 50 ns molecular dynamics simulation was conducted for saquinavir, ritonavir and remdesivir to evaluate the stability of these drugs inside the binding pocket of SARS-CoV-2 main protease. The current study provides a powerful in silico results, means for rapid screening of drugs as anti-protease medications and recommend that the above-mentioned drugs can be used in the treatment of SARS-CoV-2 in combined or sole therapy.Communicated by Ramaswamy H. Sarma.
Keywords: MD simulation; Mpro; PCA; SARS-CoV-2; covalent docking; drug repurposing.
Figures
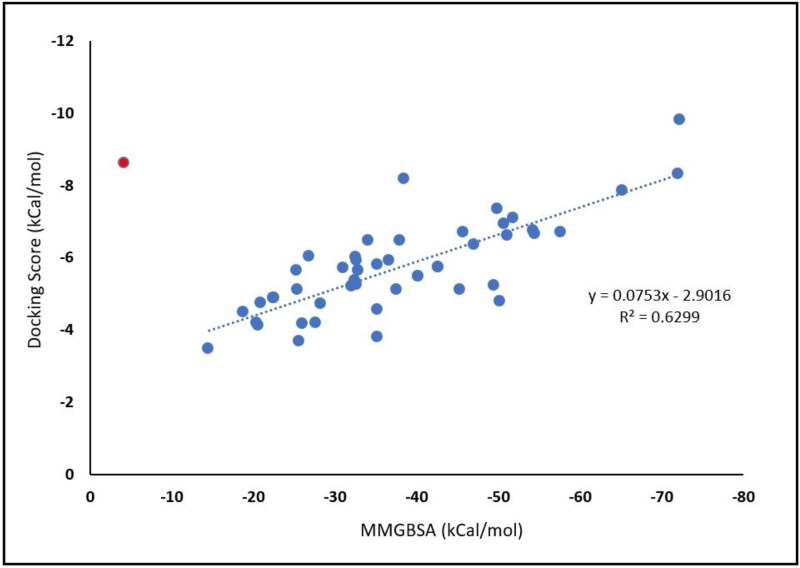
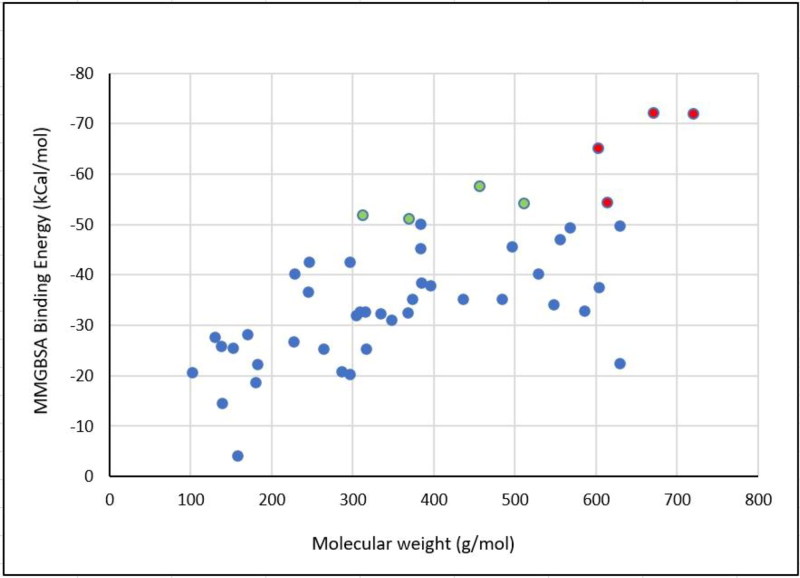
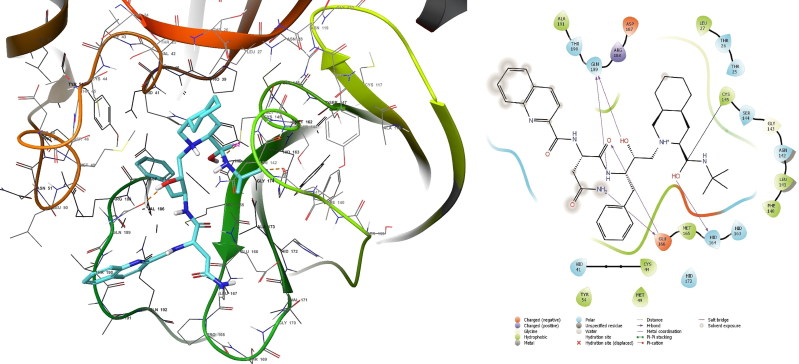
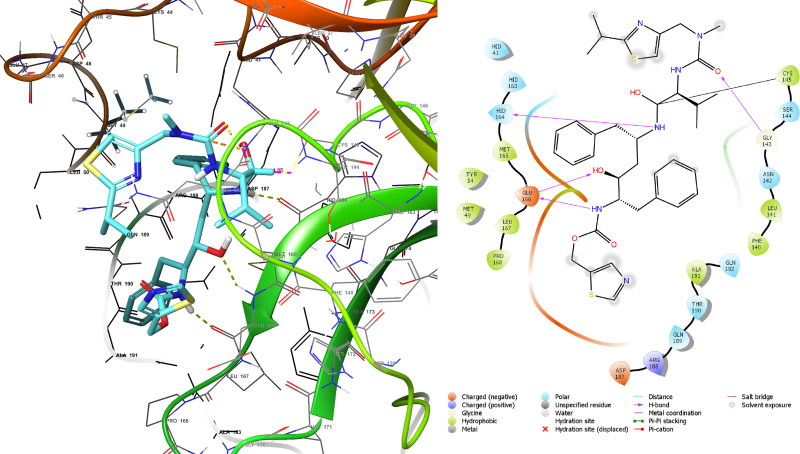
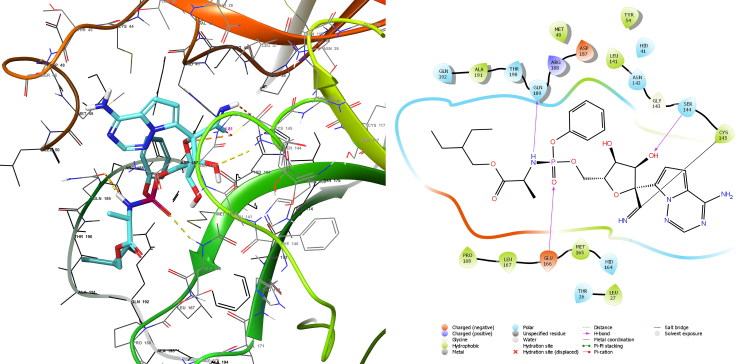
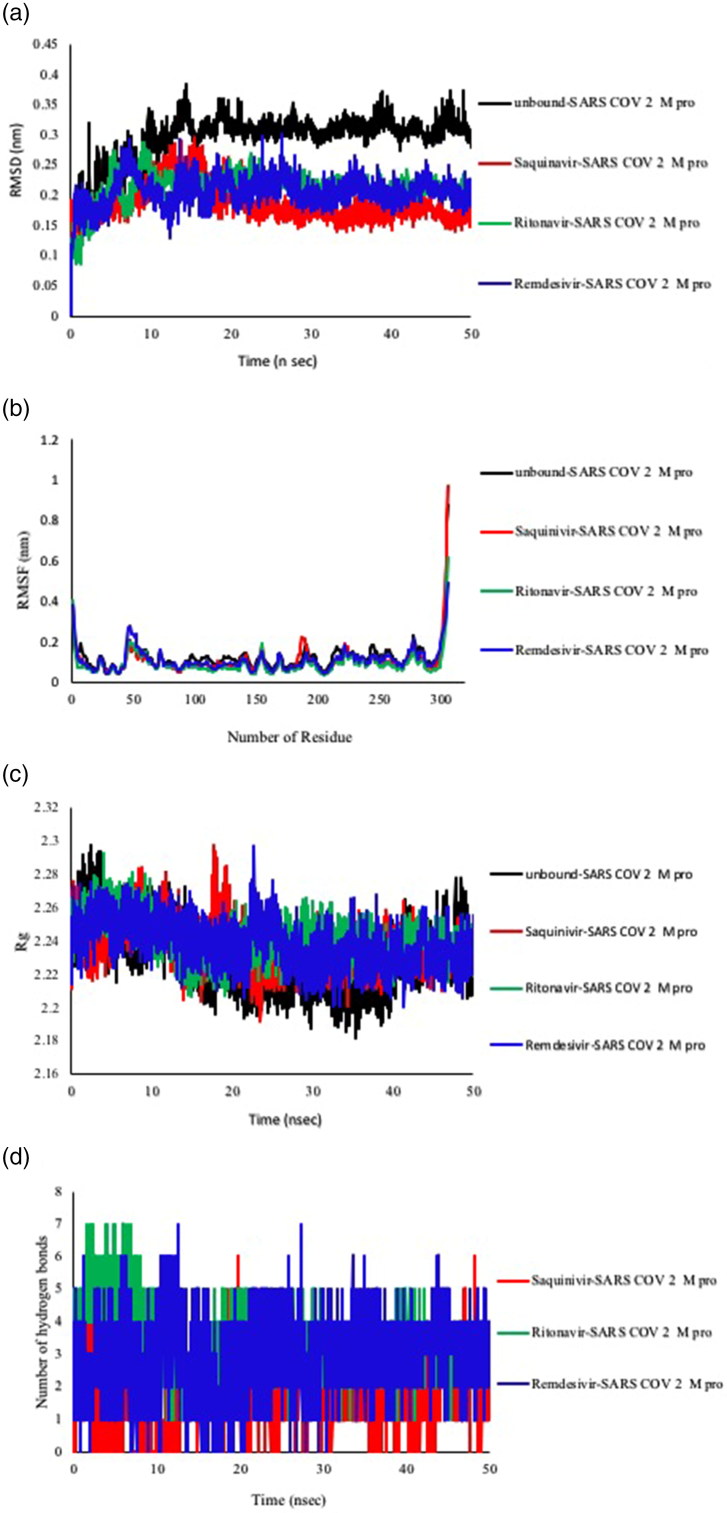
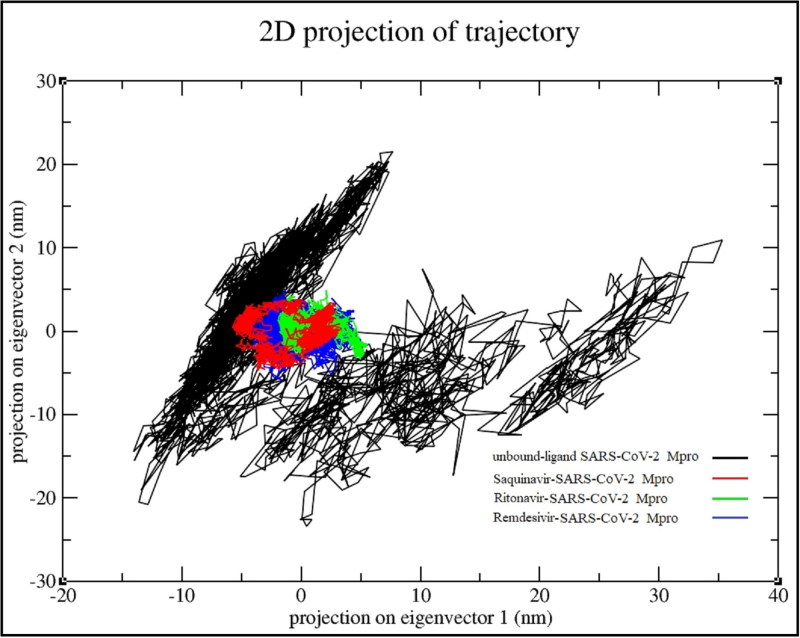
References
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., & Lindahl E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1-2, 19–25. 10.1016/j.softx.2015.06.001 - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous