

Sulfonate N-Heterocyclic Carbene-Copper Complexes: Uniquely Effective Catalysts for Enantioselective Synthesis of C-C, C-B, C-H, and C-Si Bonds
- PMID: 32364640
- PMCID: PMC7609658
- DOI: 10.1002/anie.202003755
Sulfonate N-Heterocyclic Carbene-Copper Complexes: Uniquely Effective Catalysts for Enantioselective Synthesis of C-C, C-B, C-H, and C-Si Bonds
Abstract
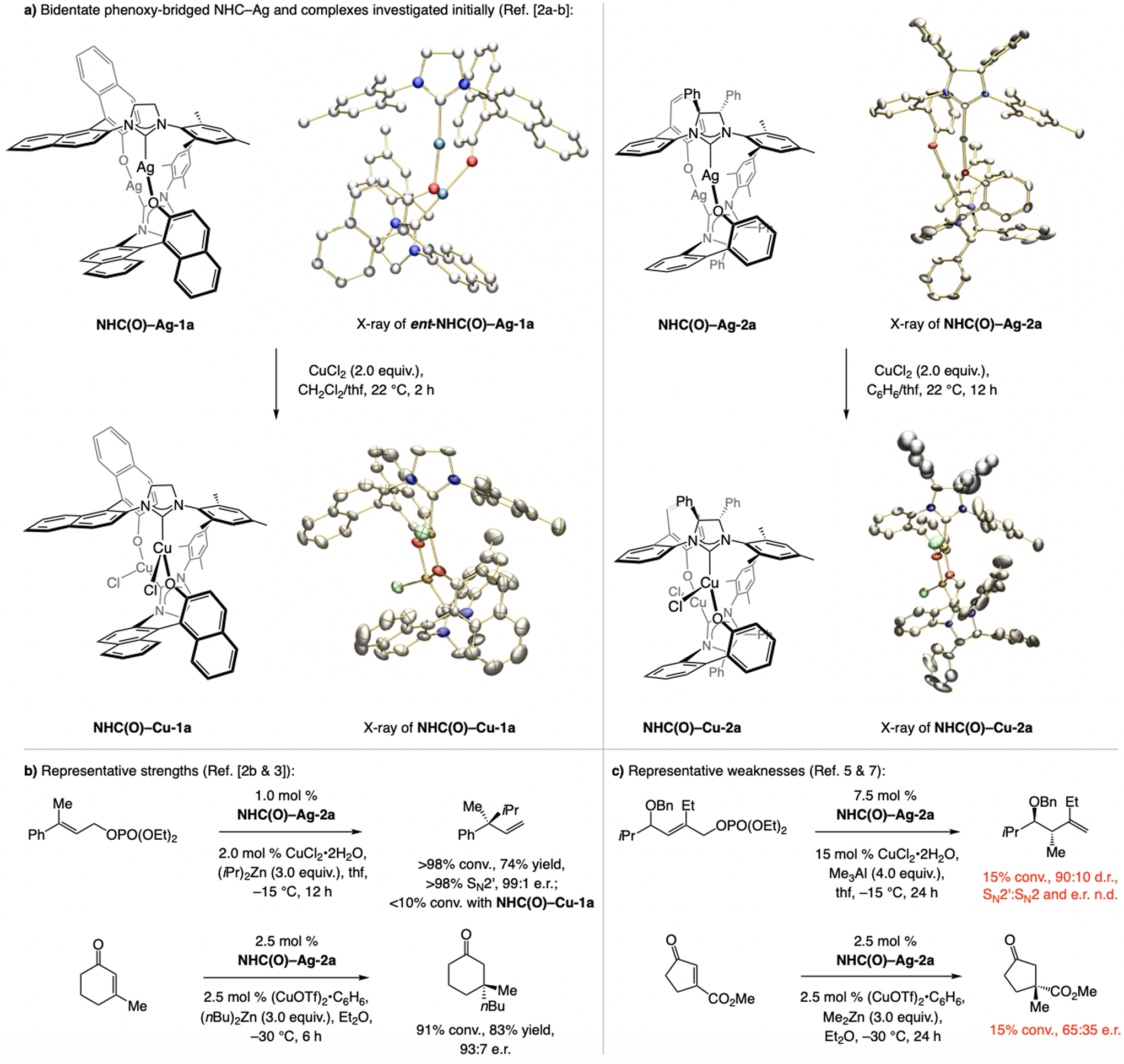
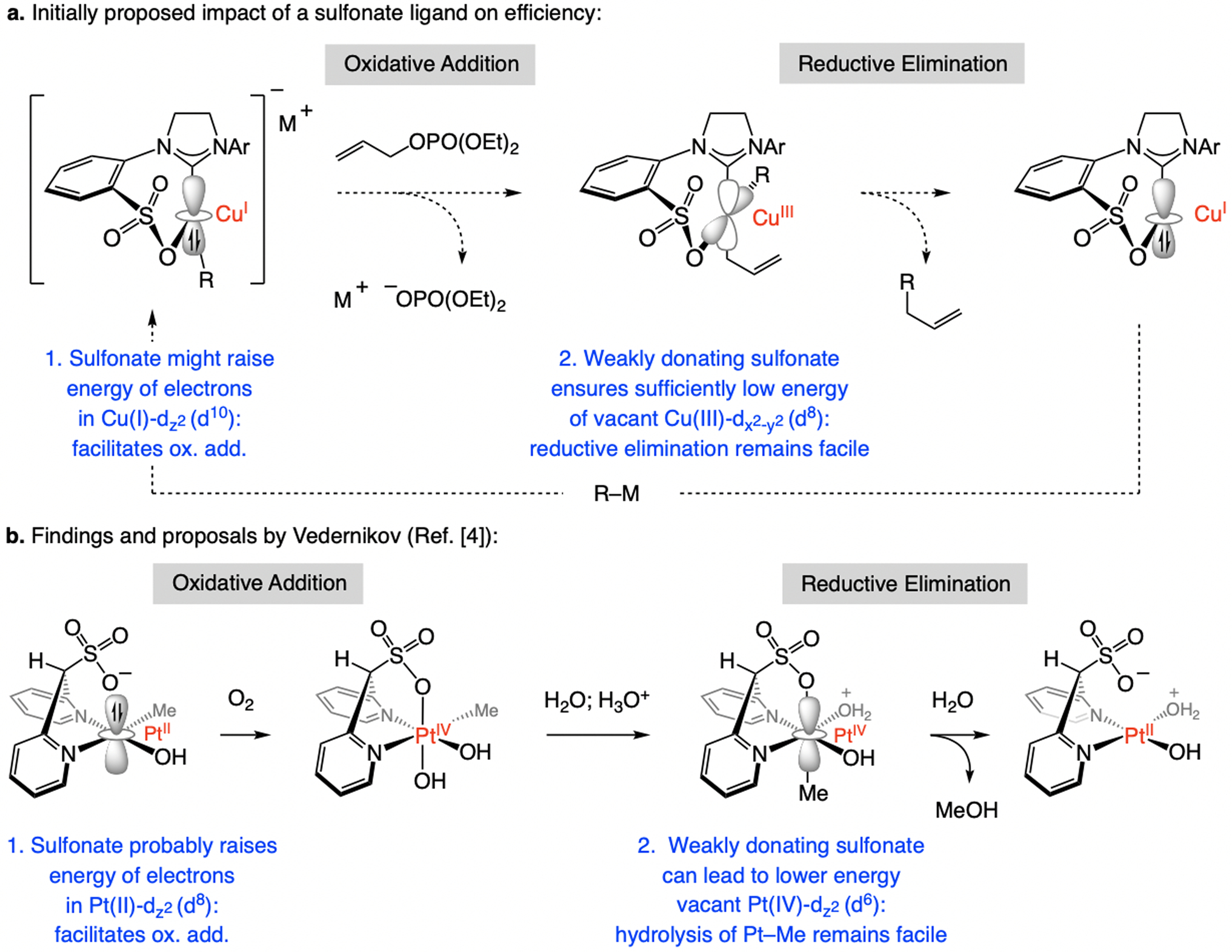
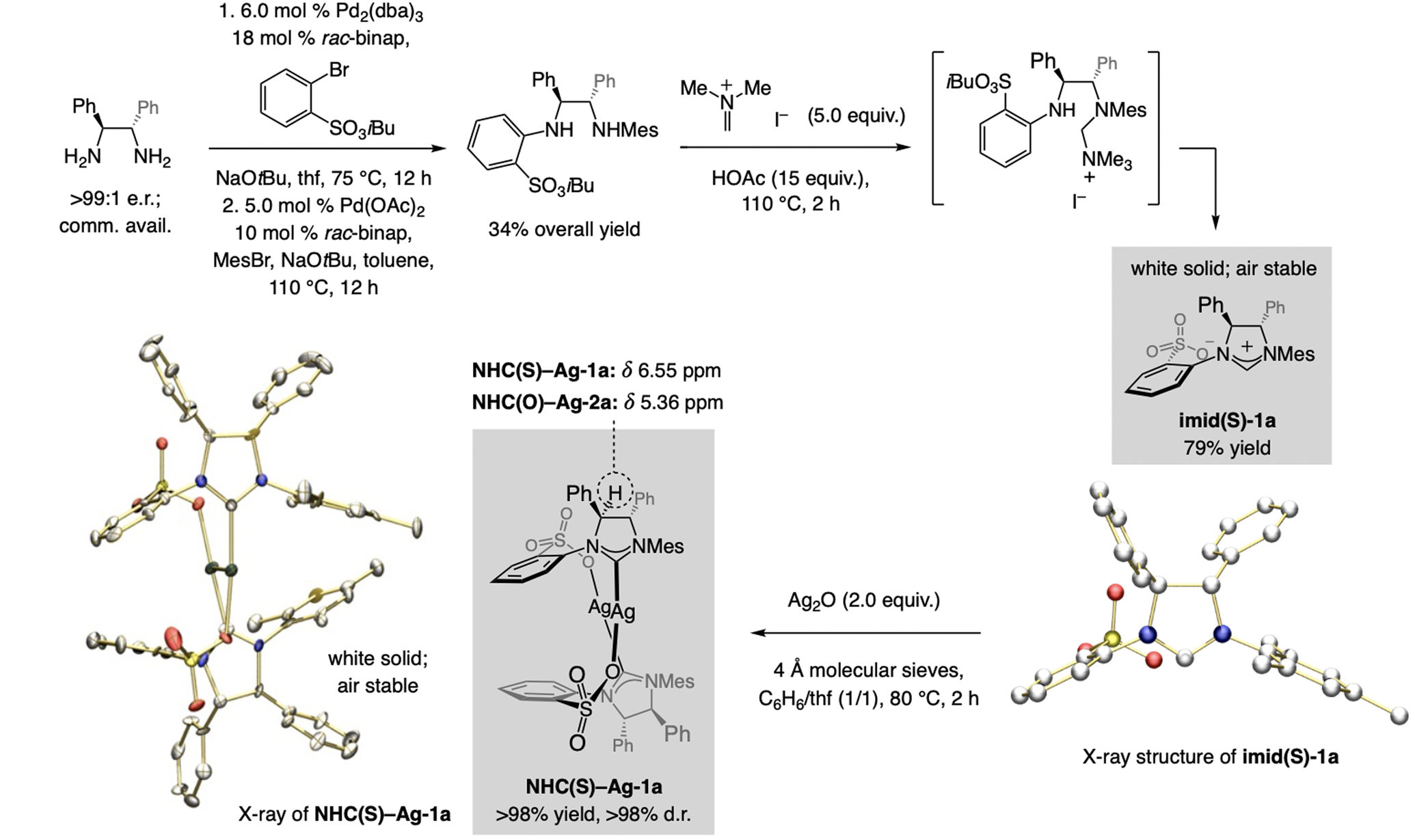
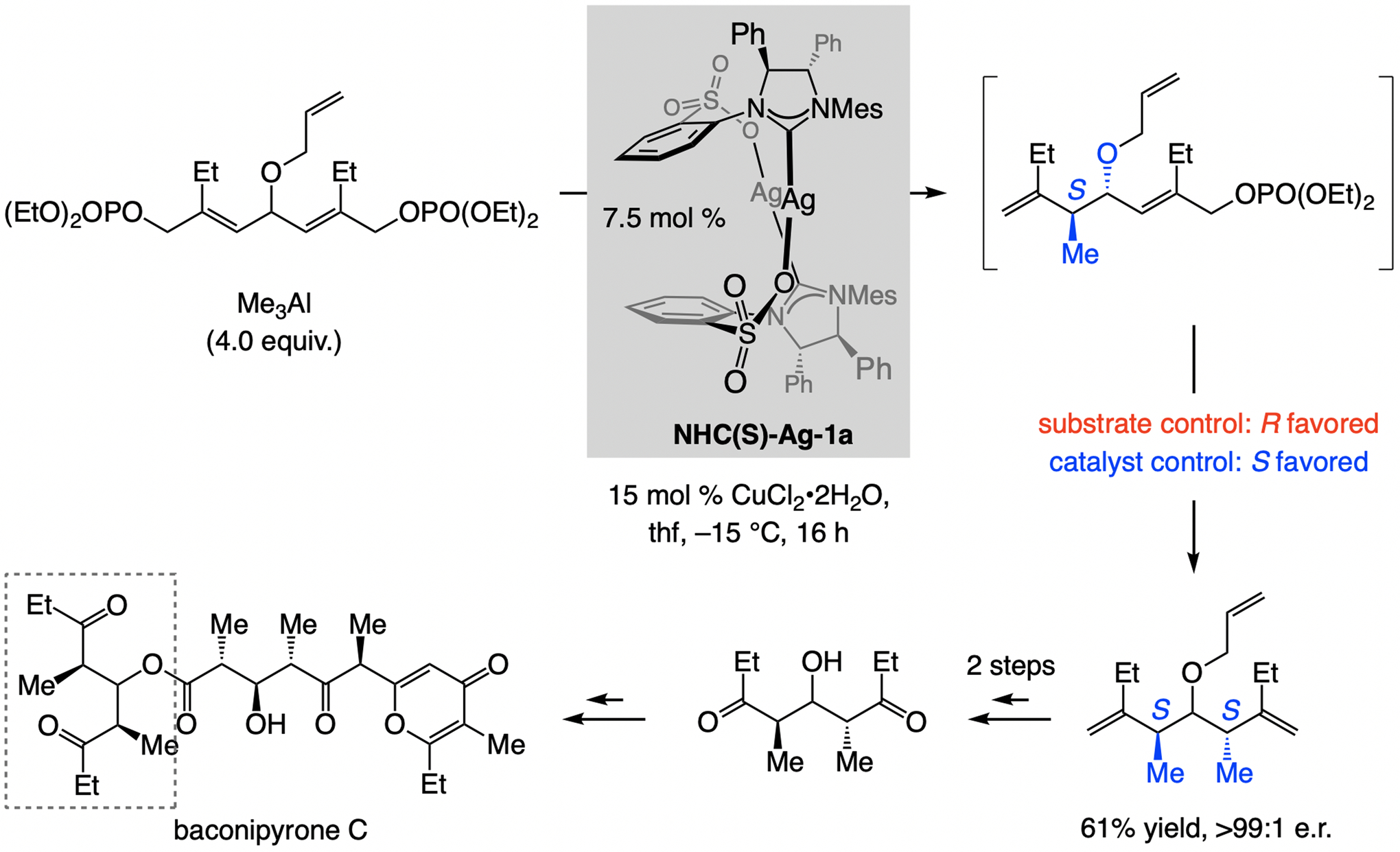
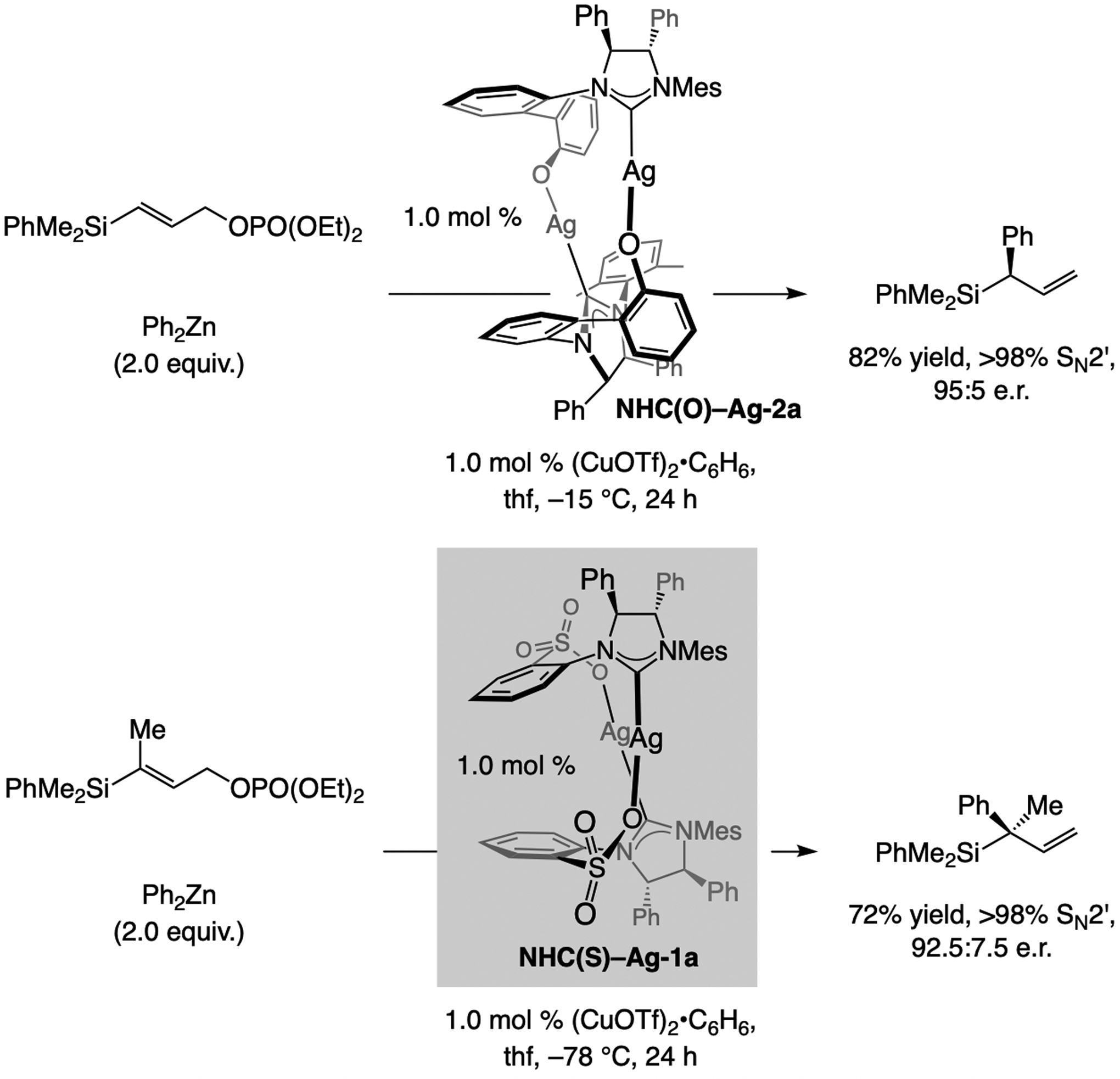
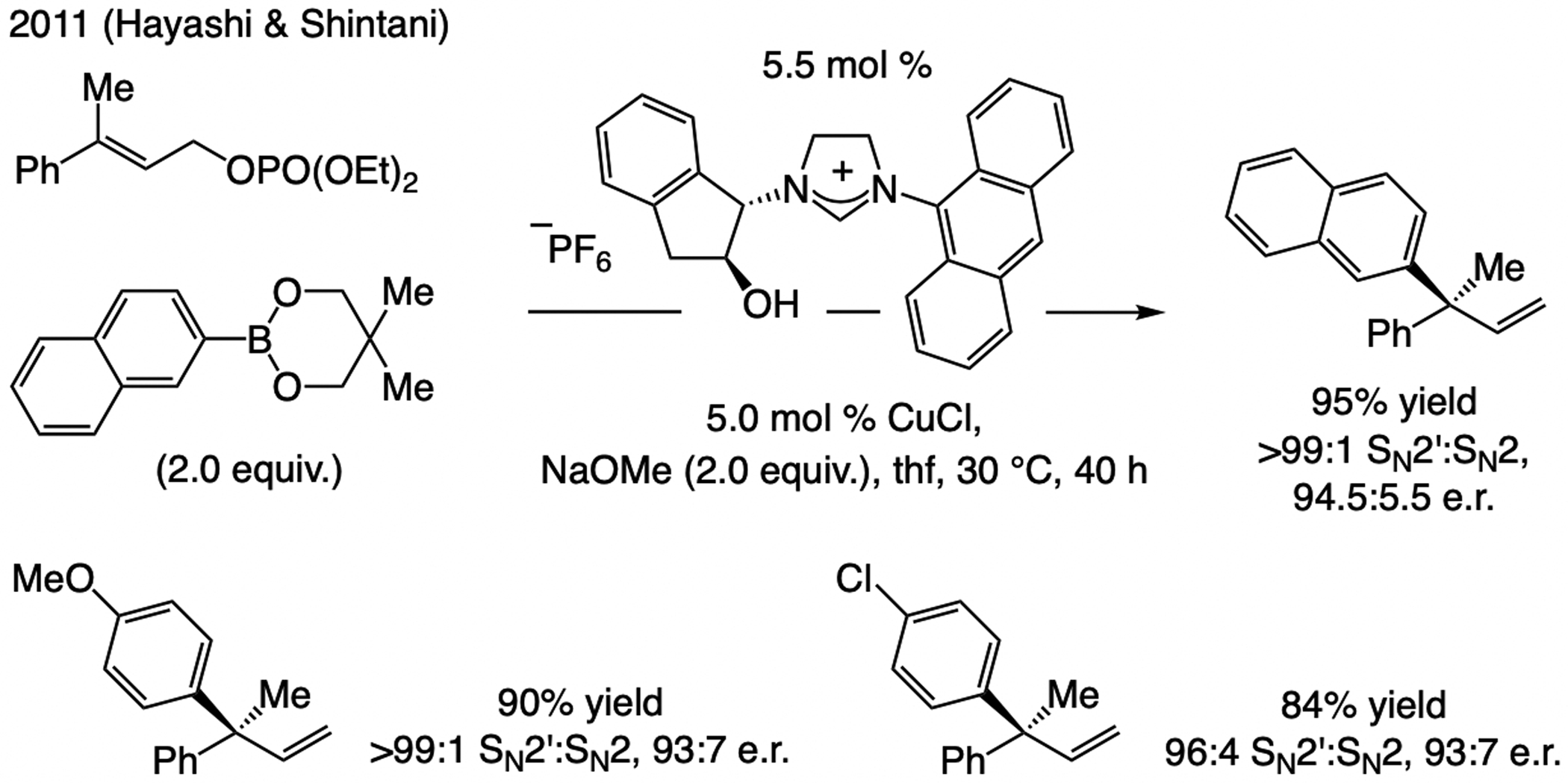
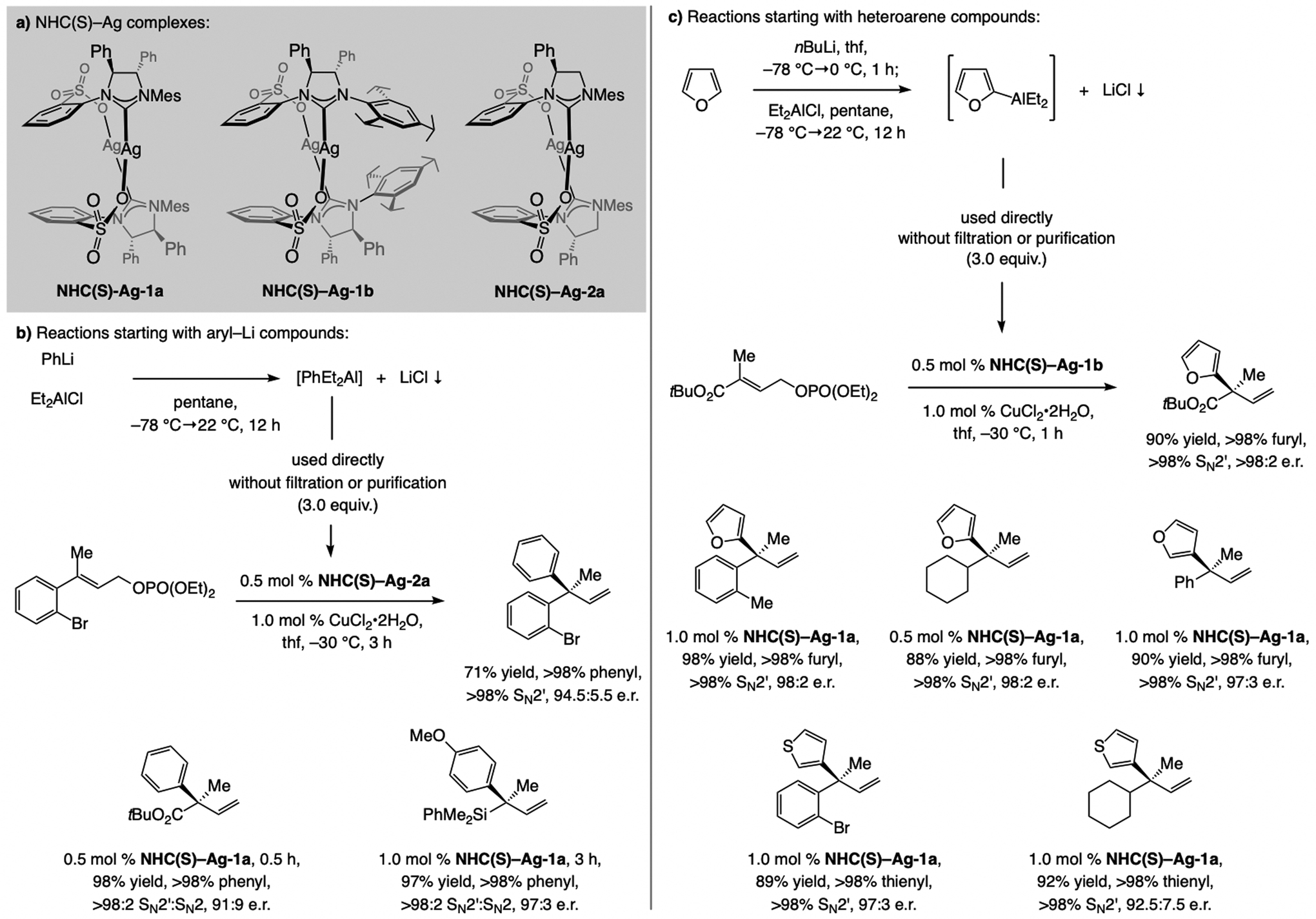
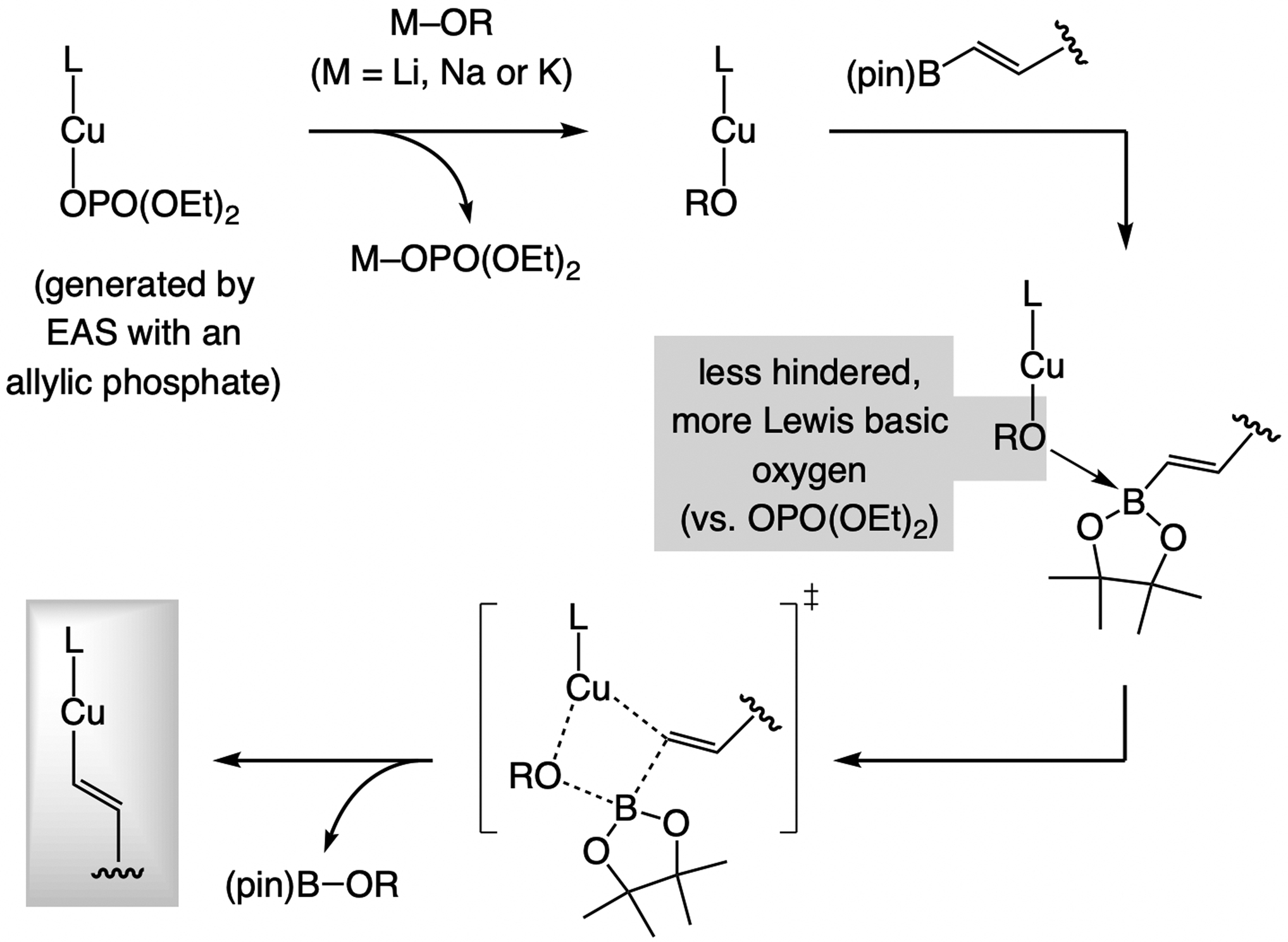
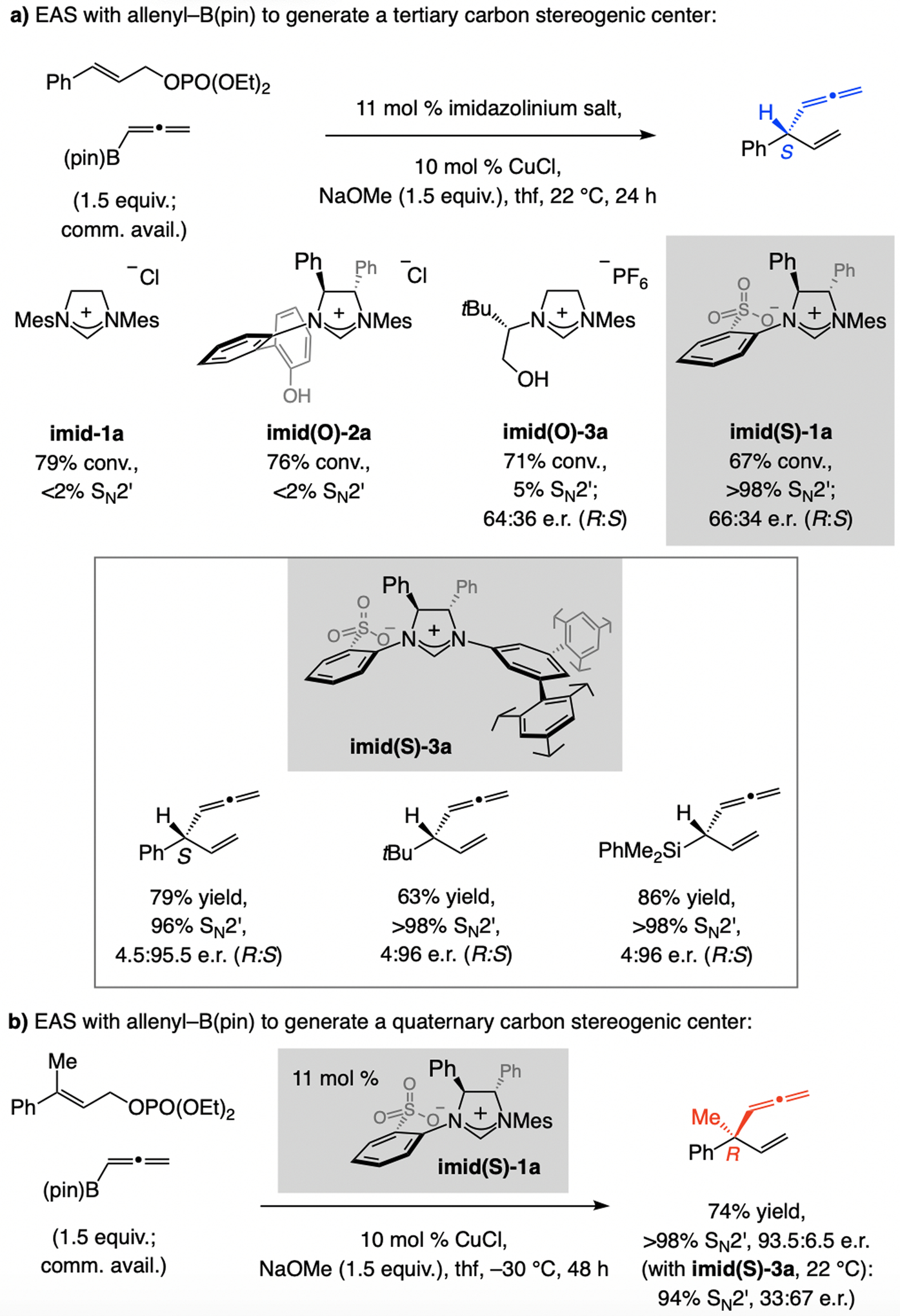
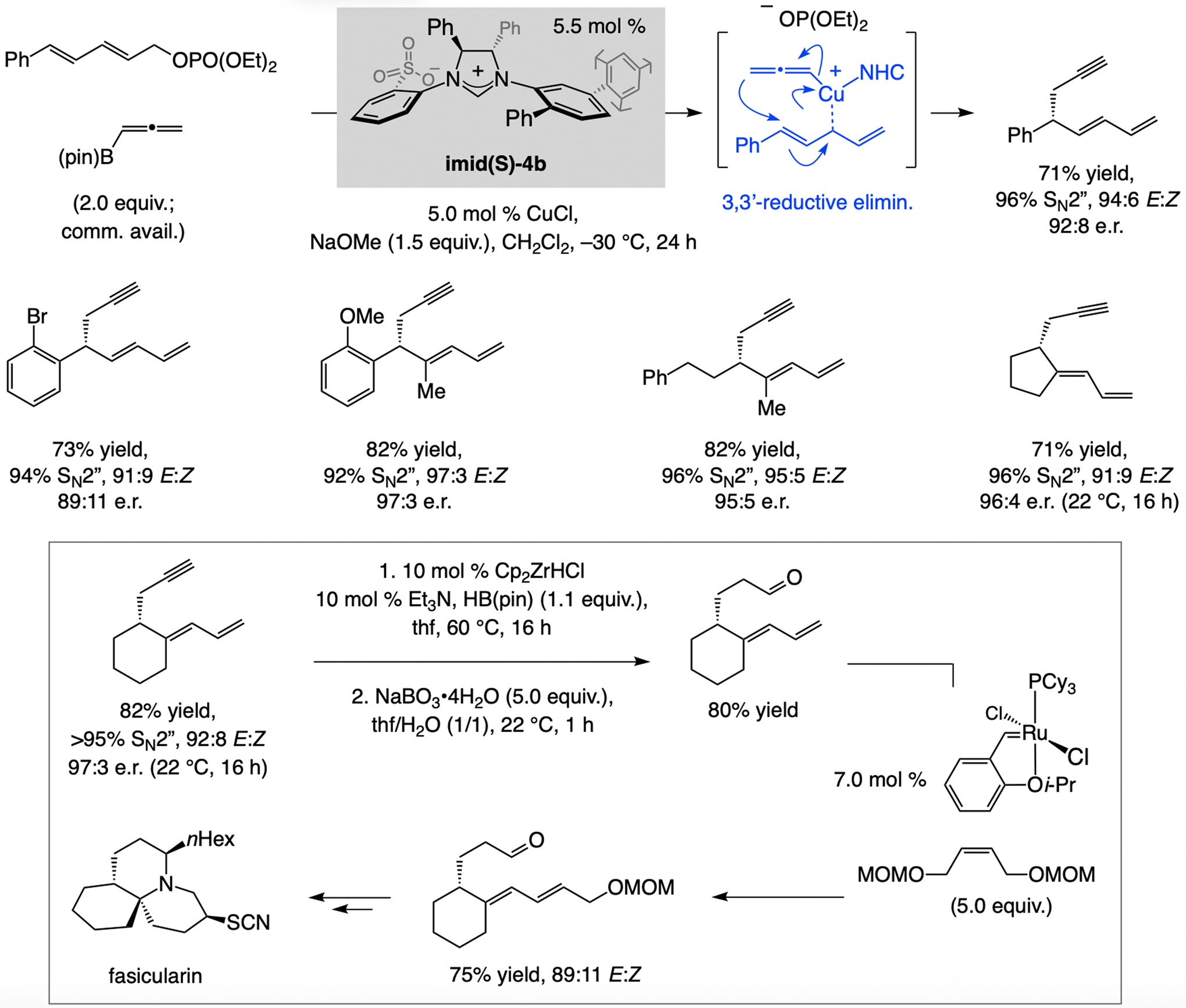
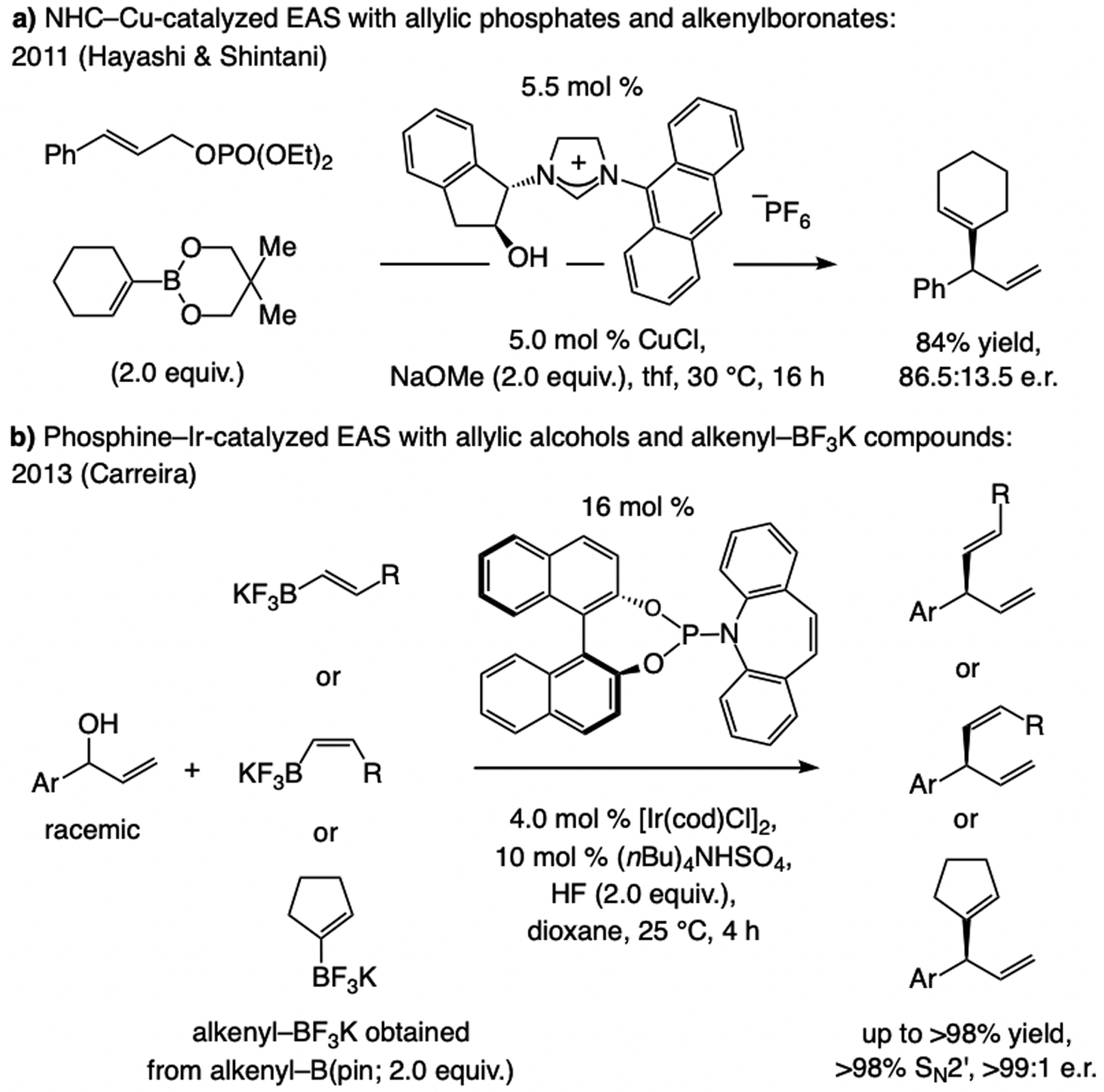
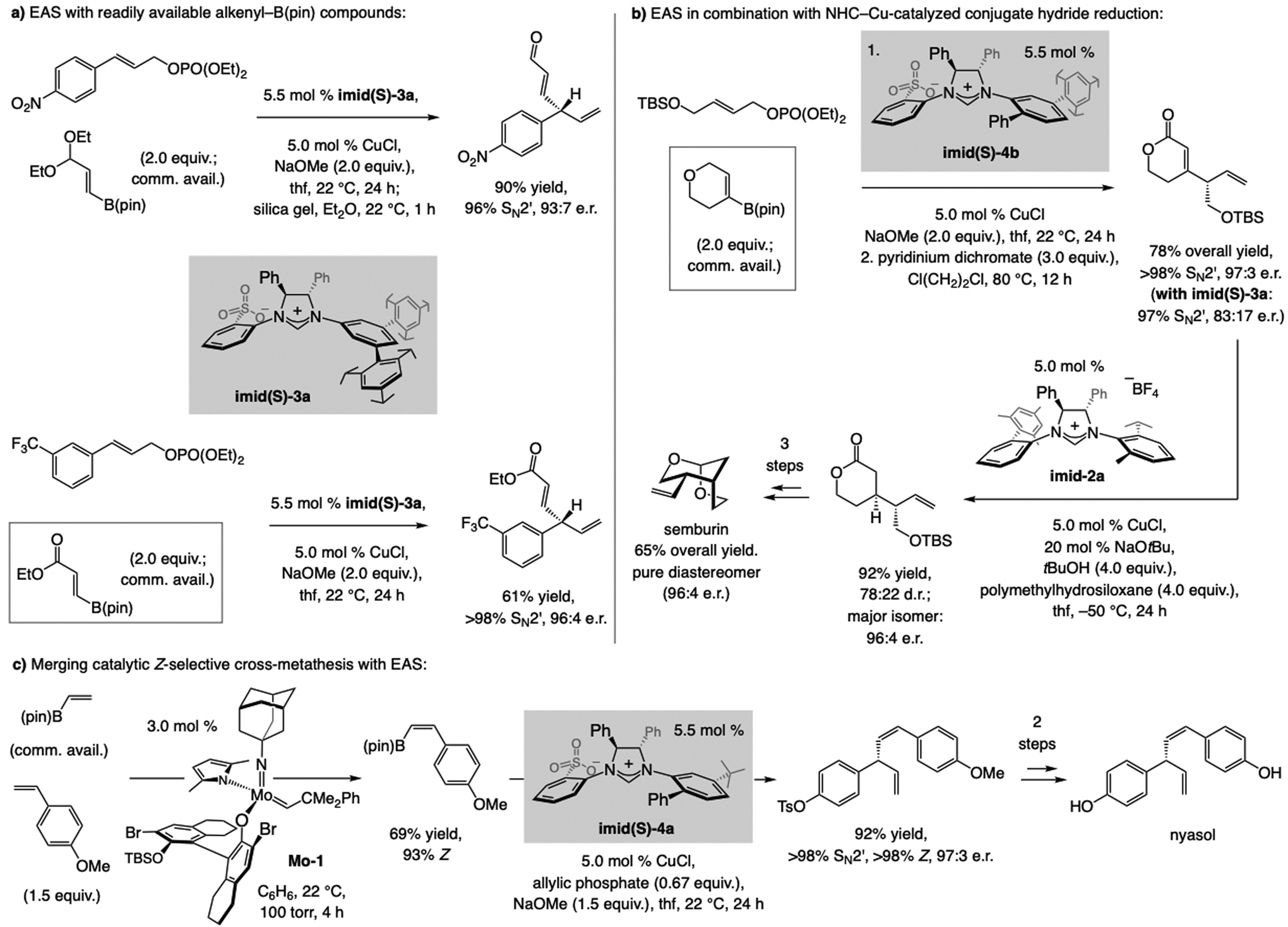
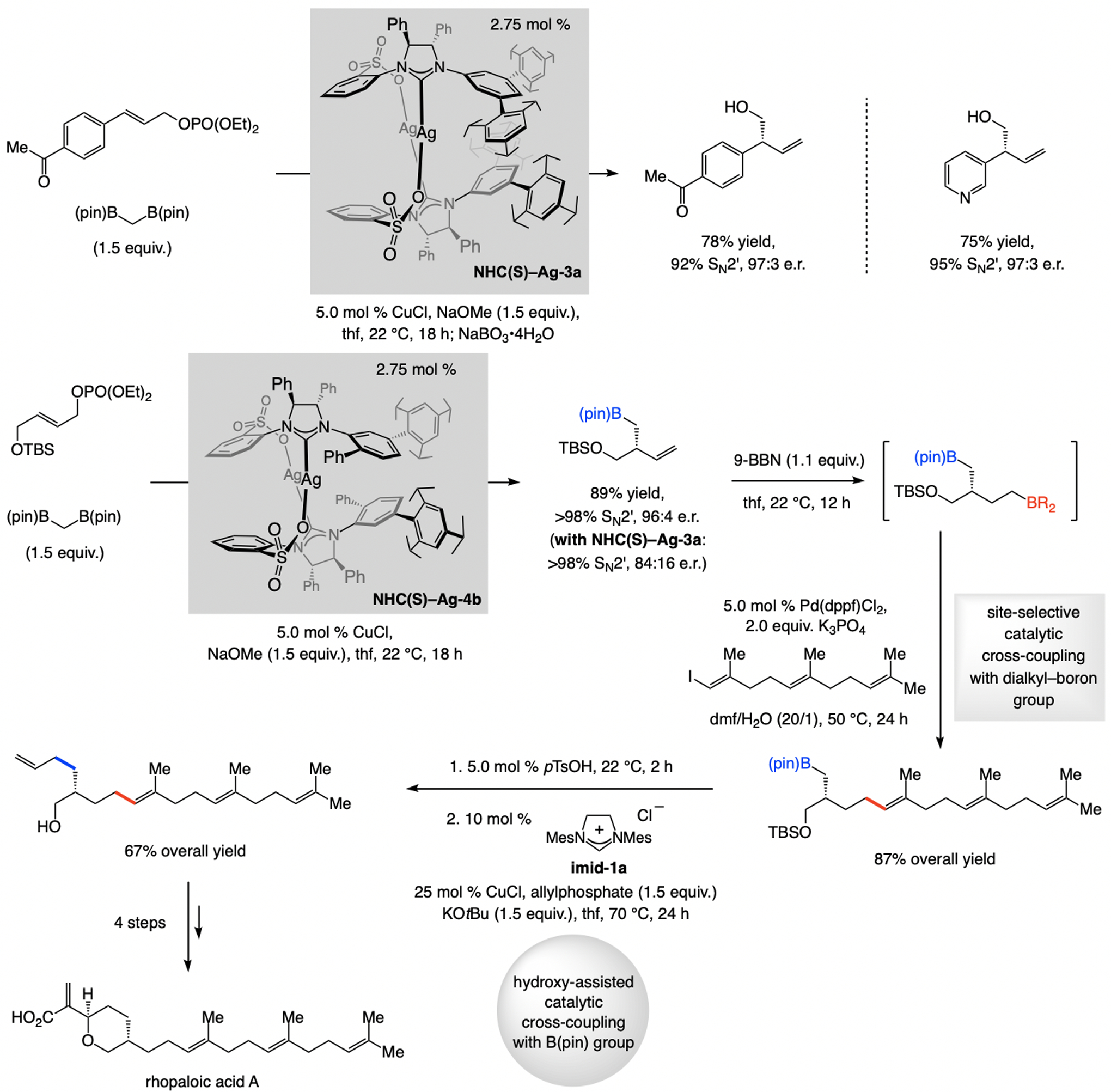
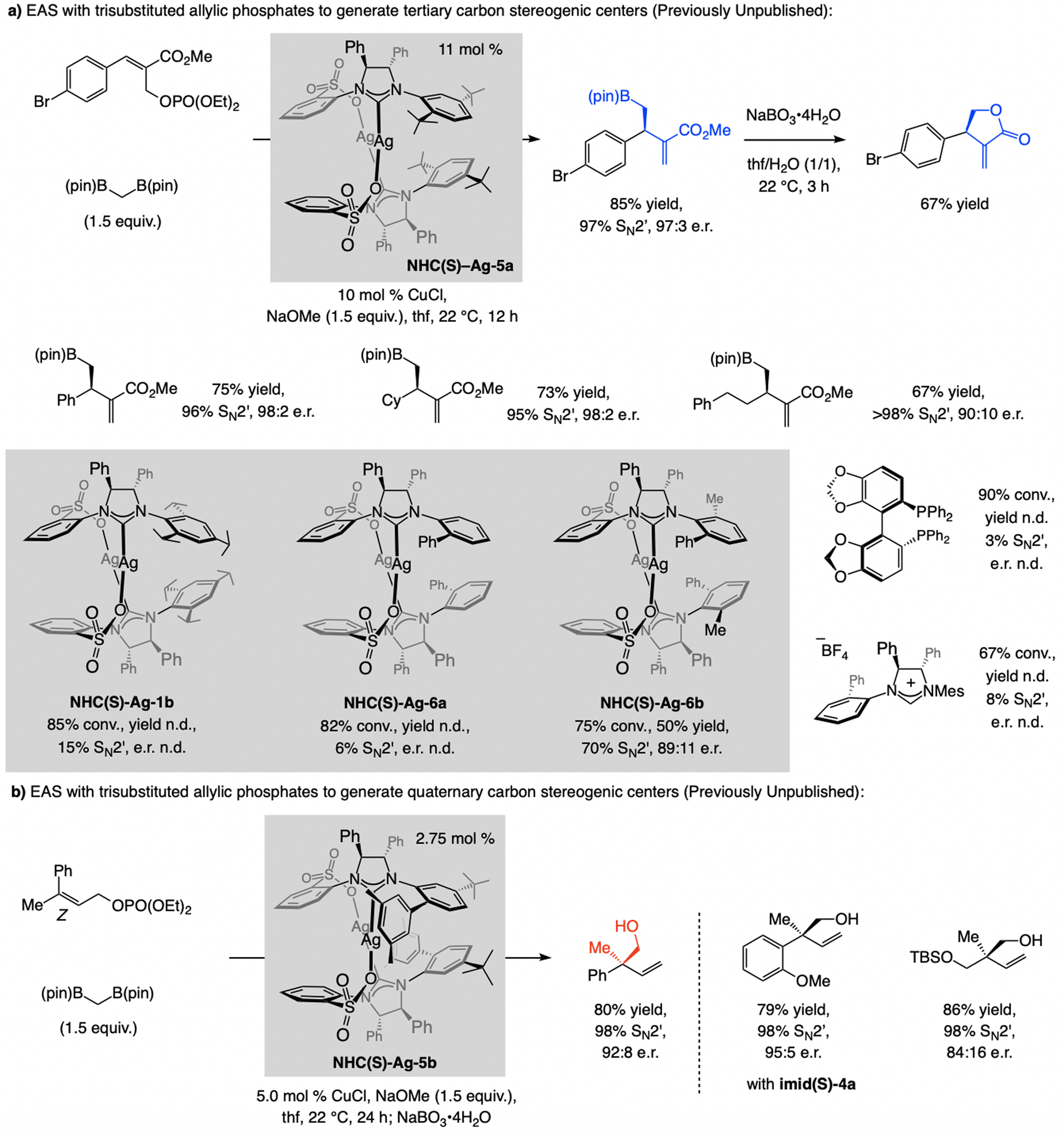
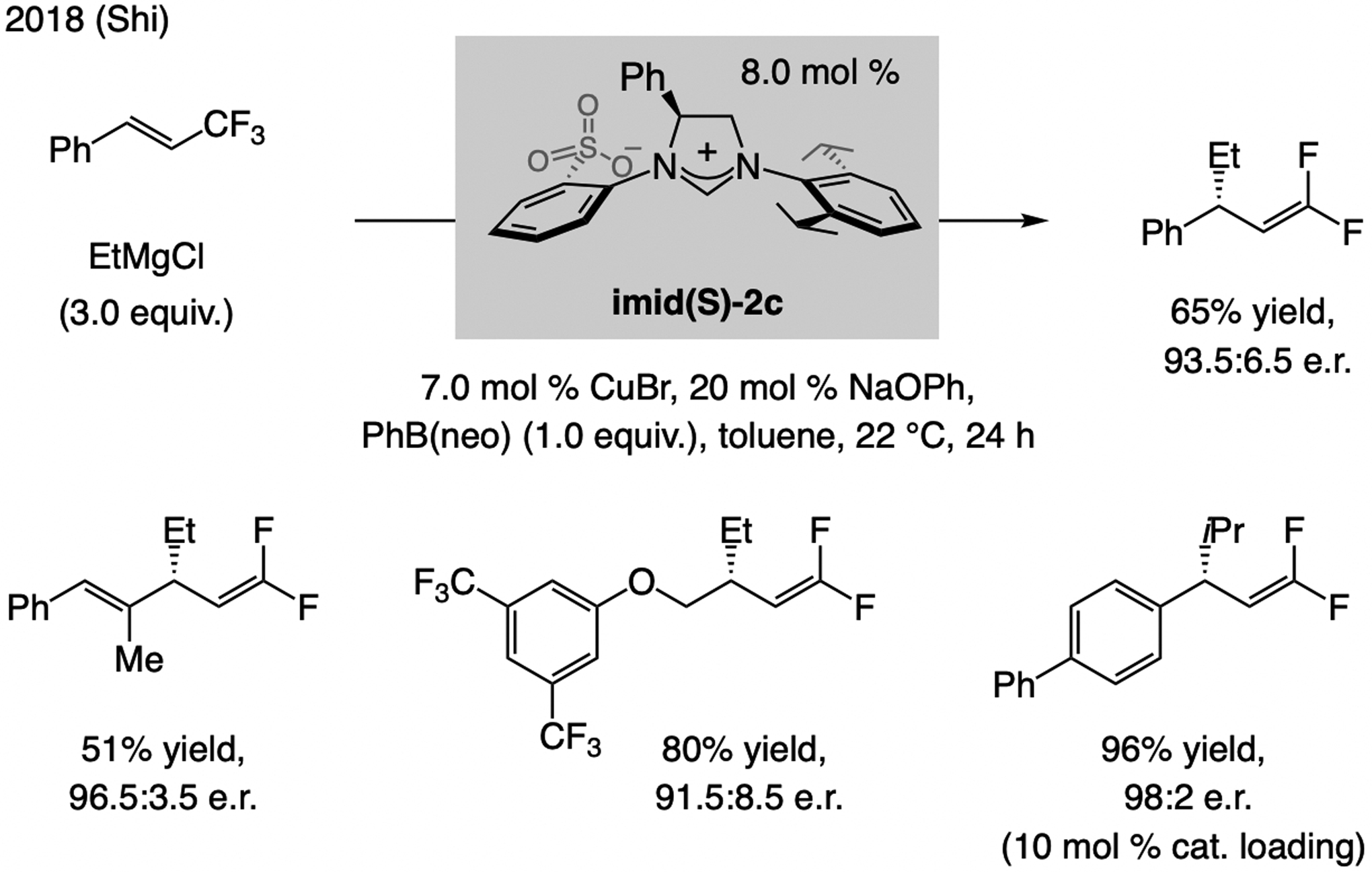
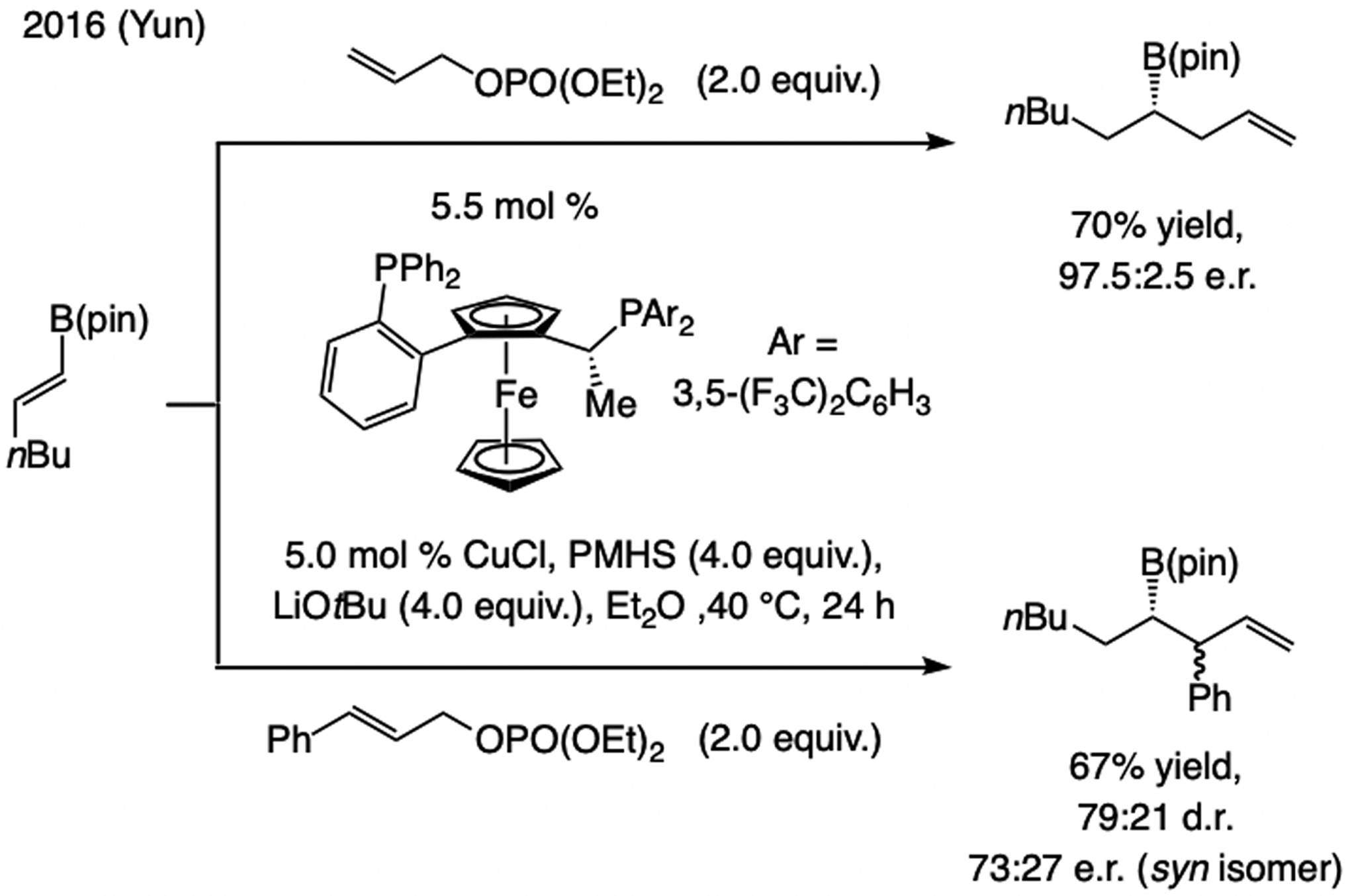
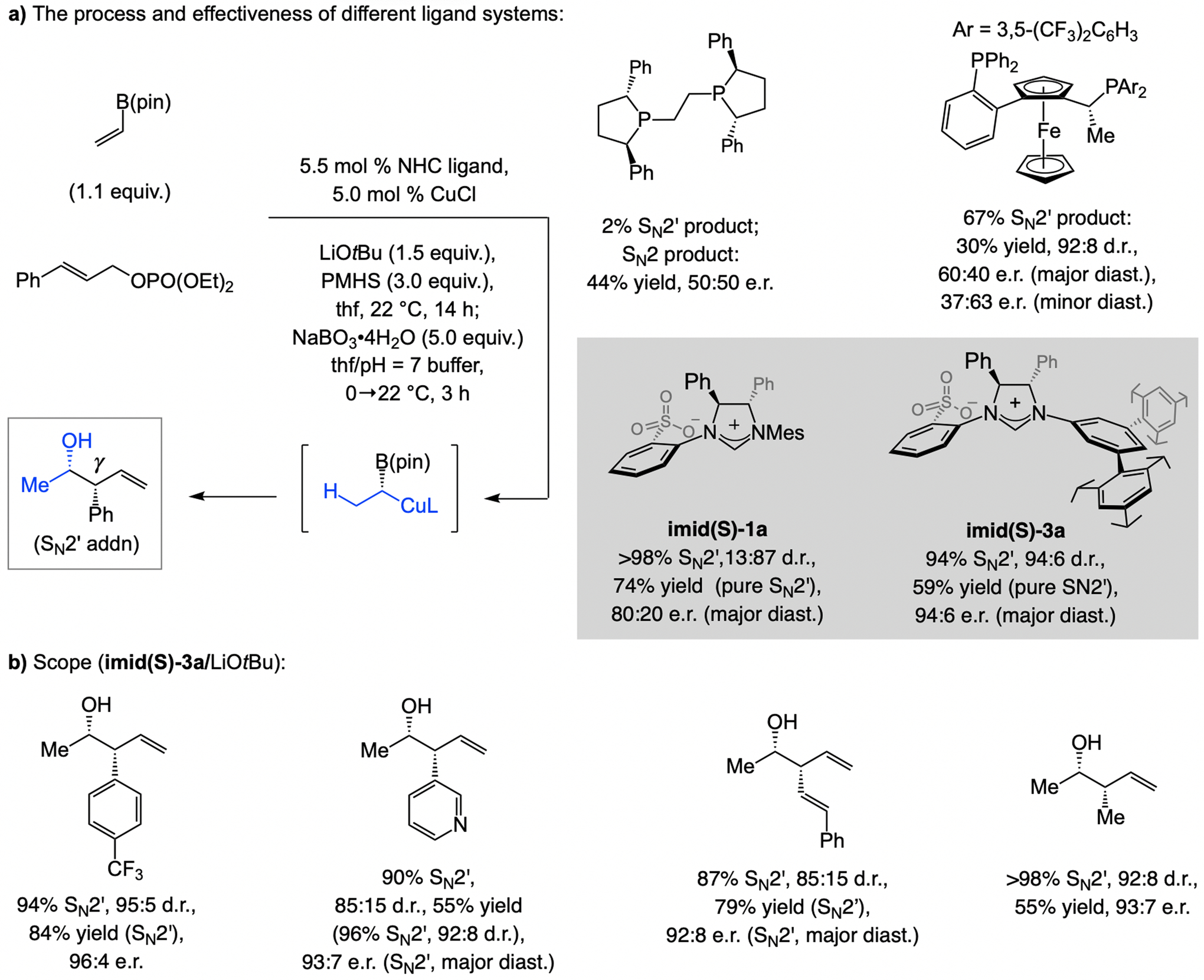
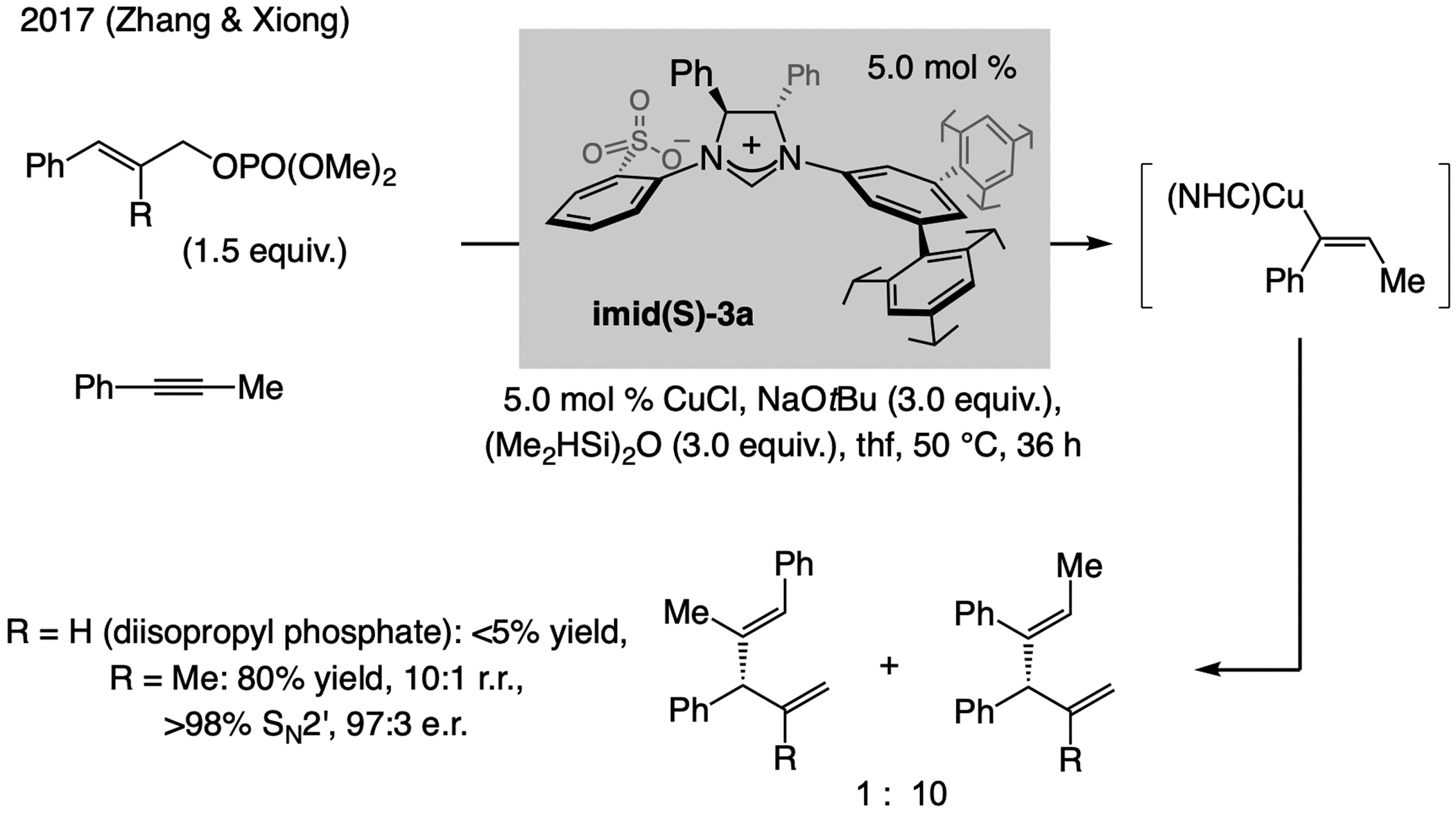
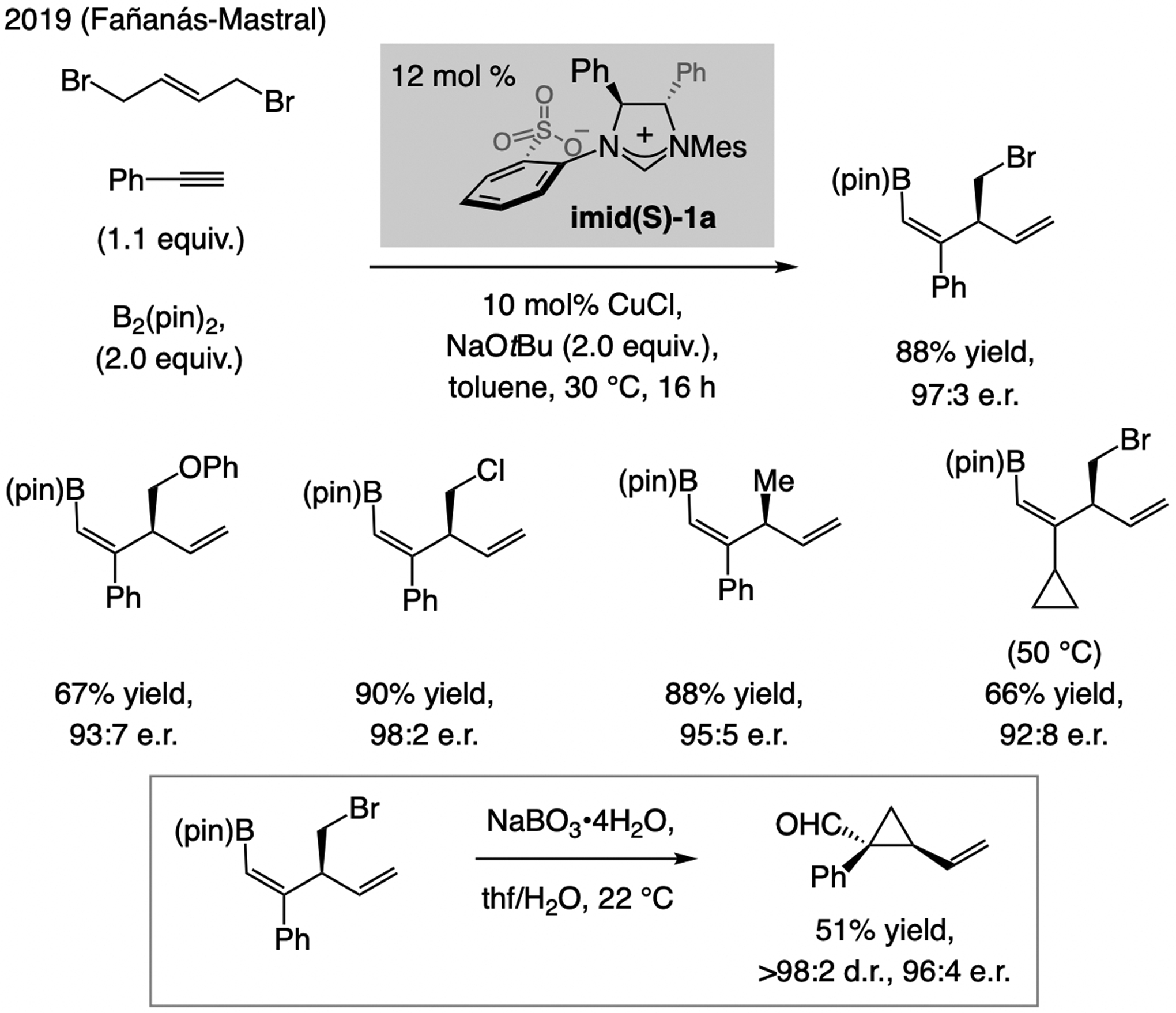
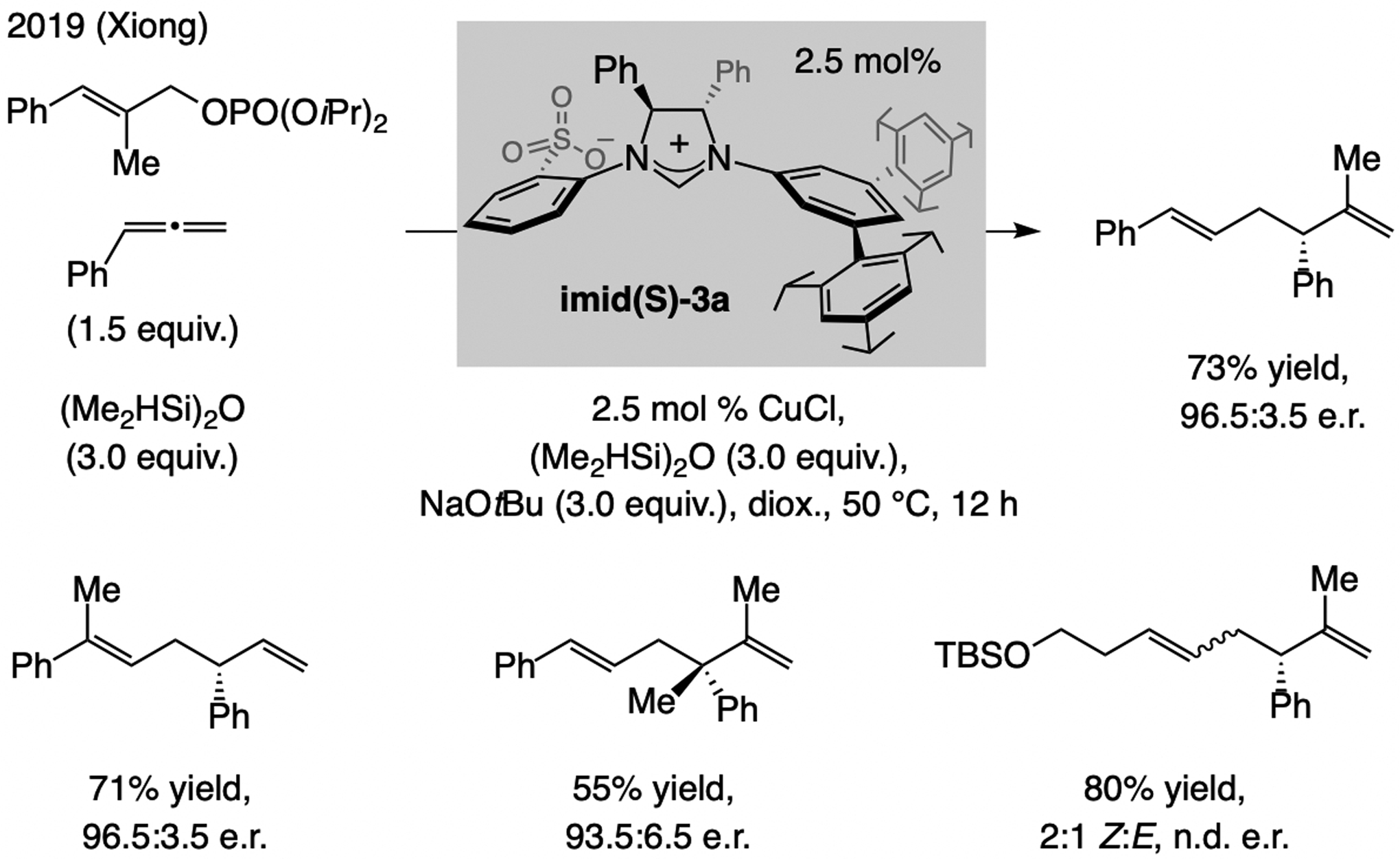
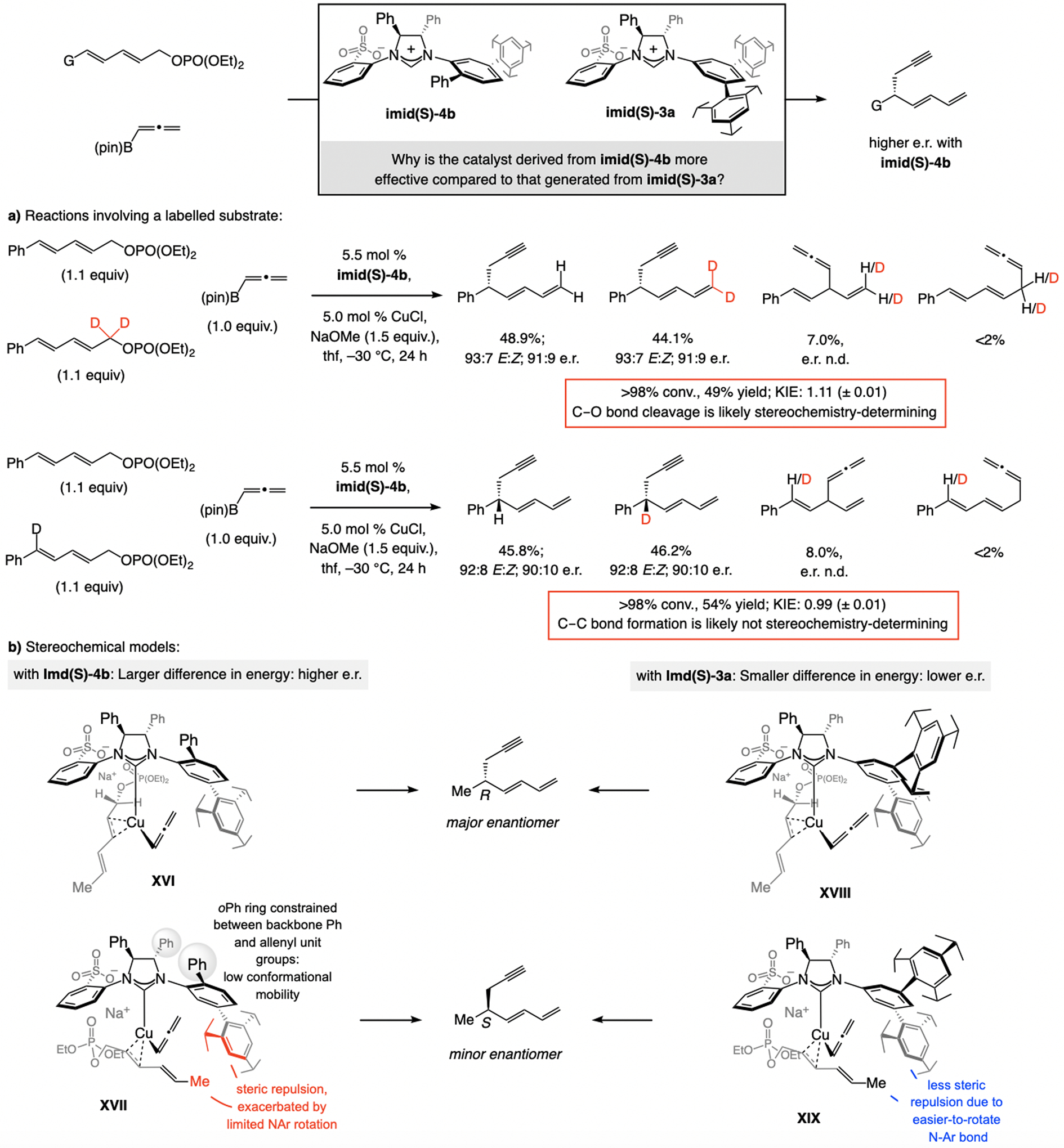
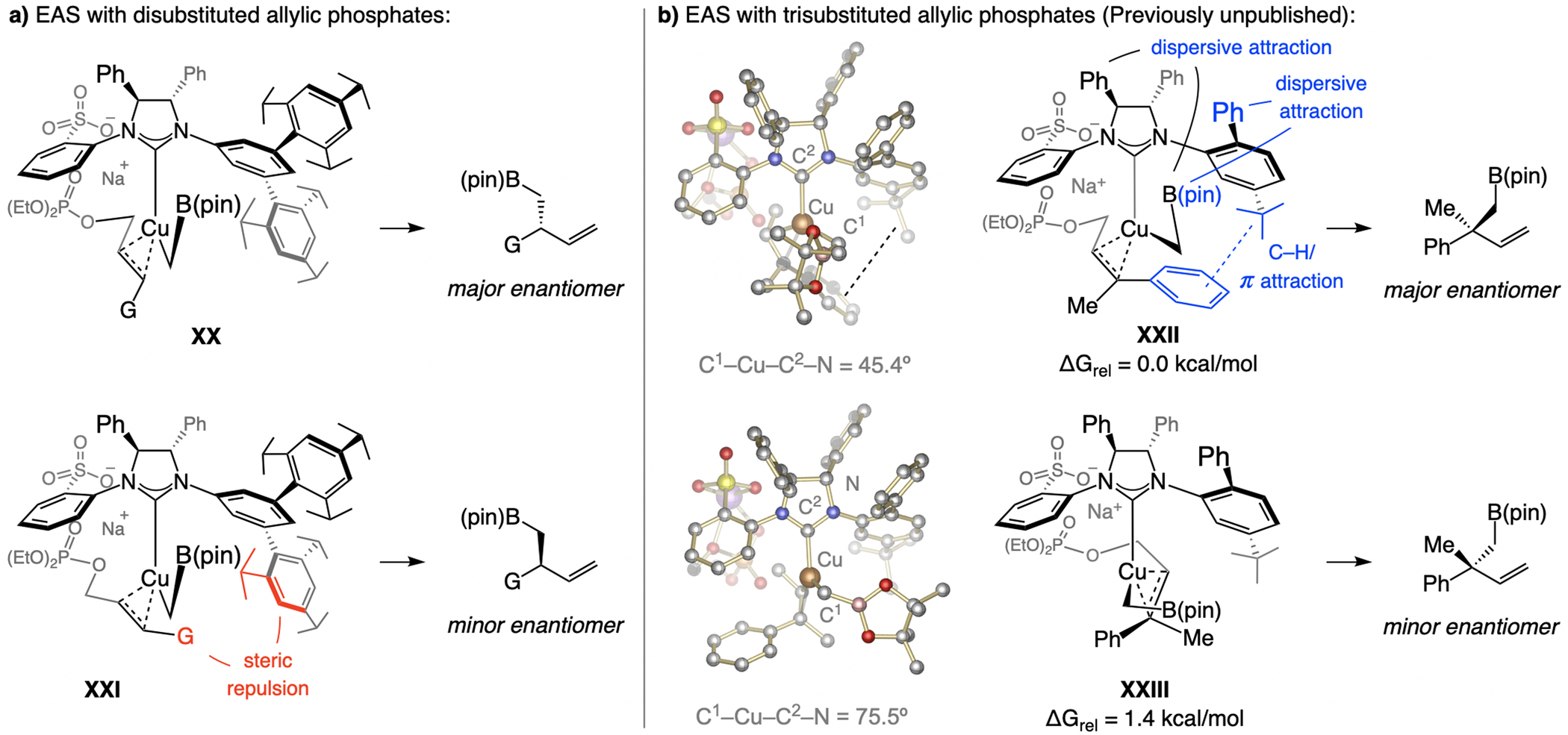
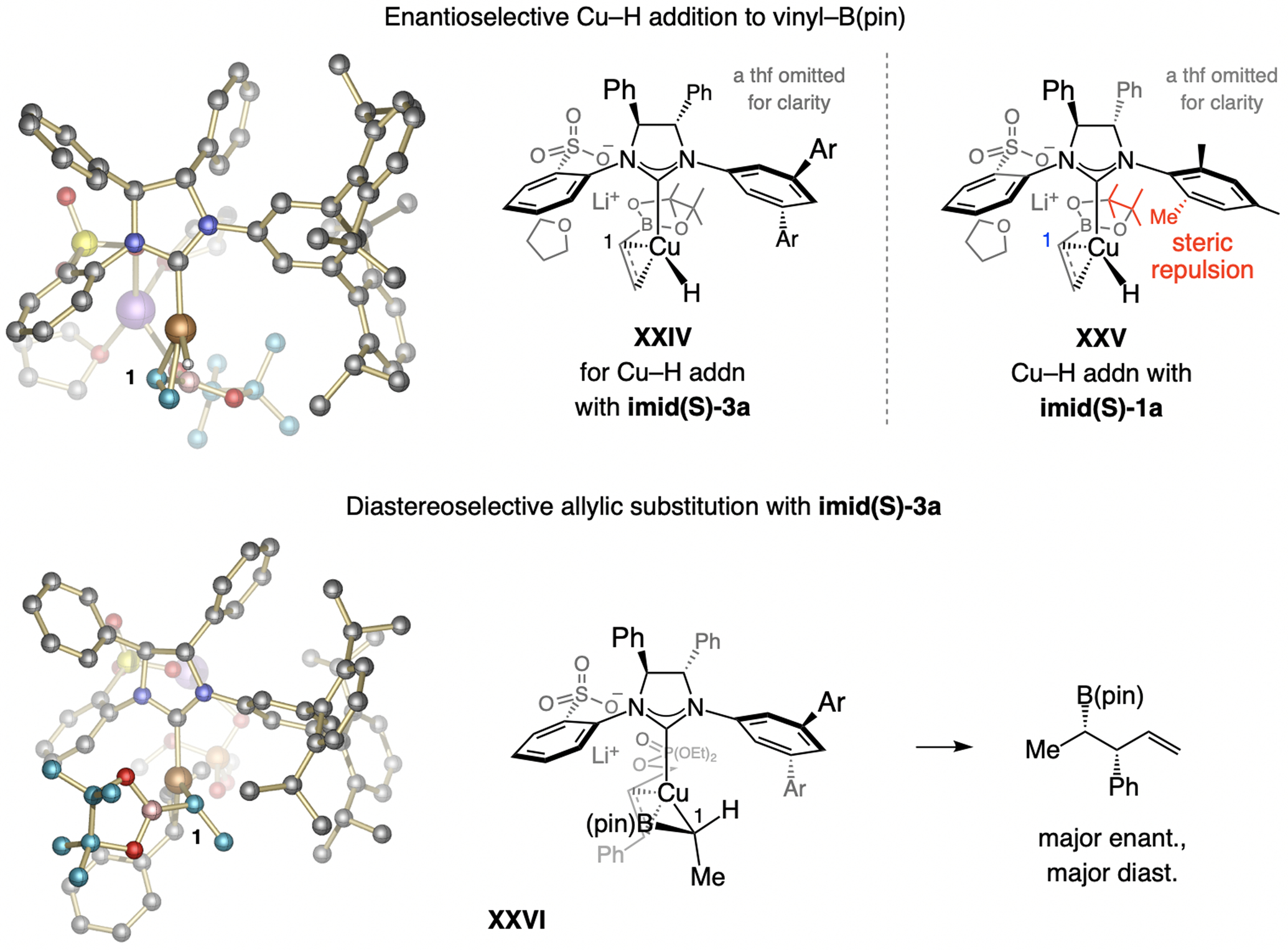
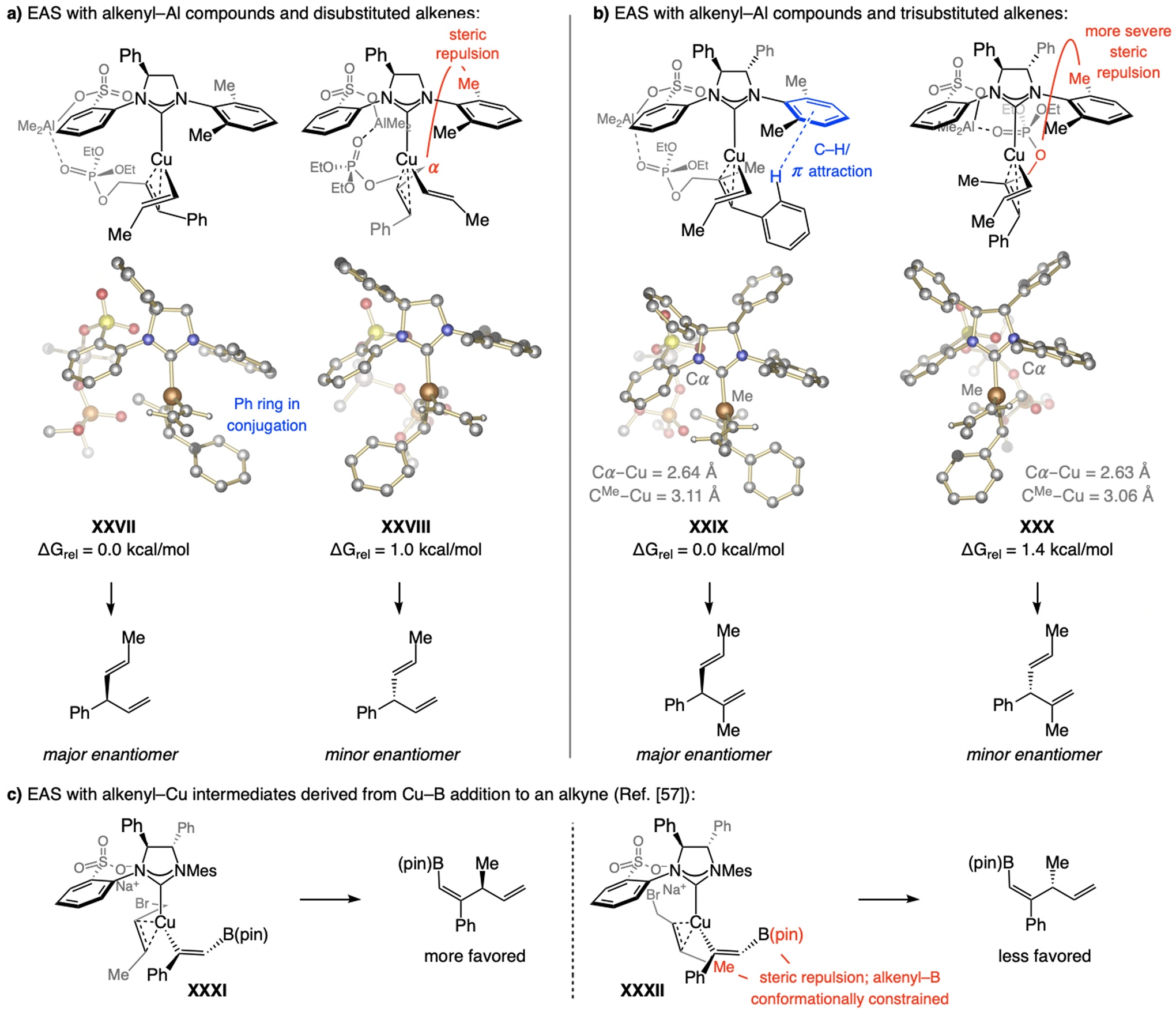
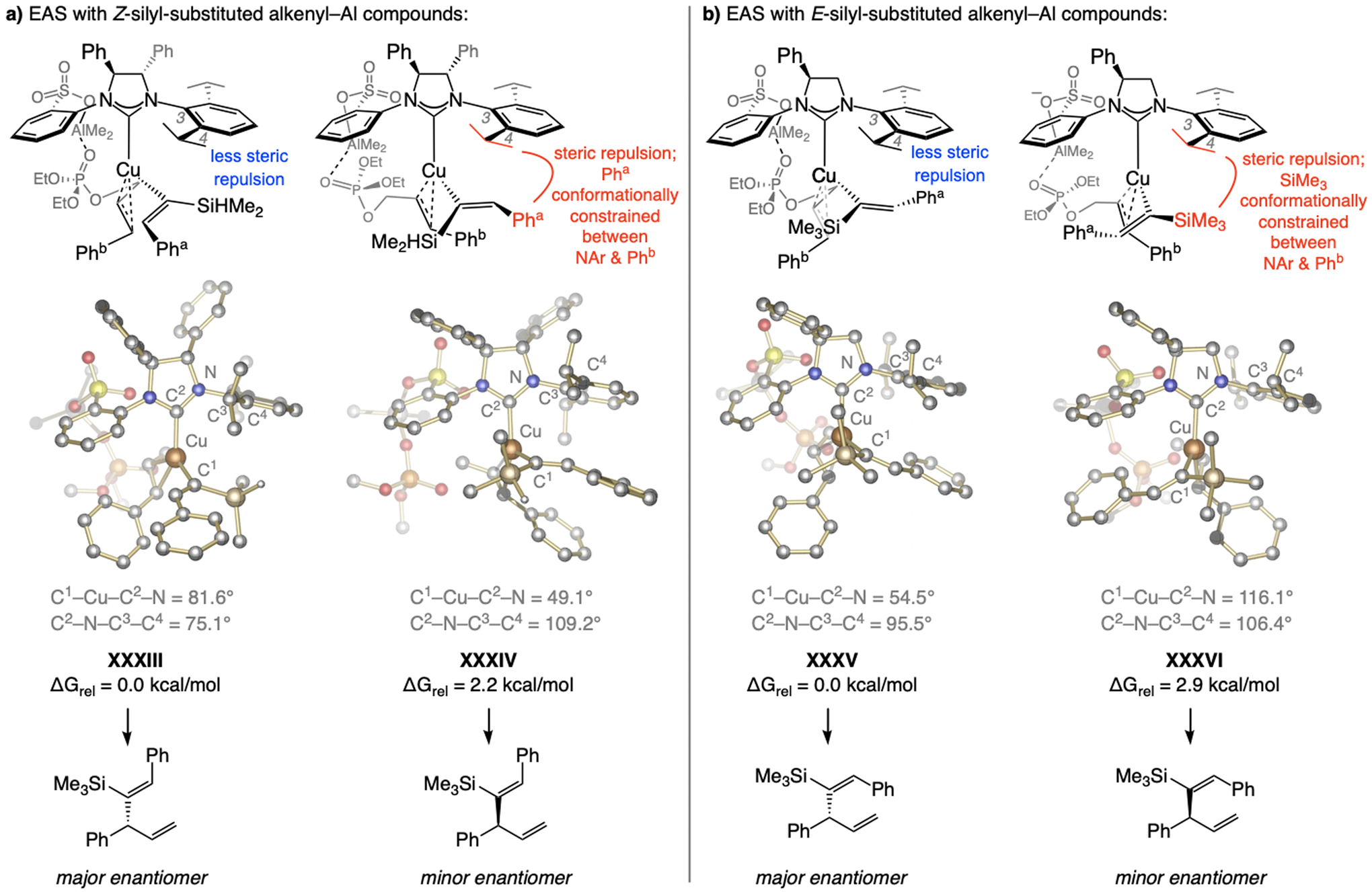
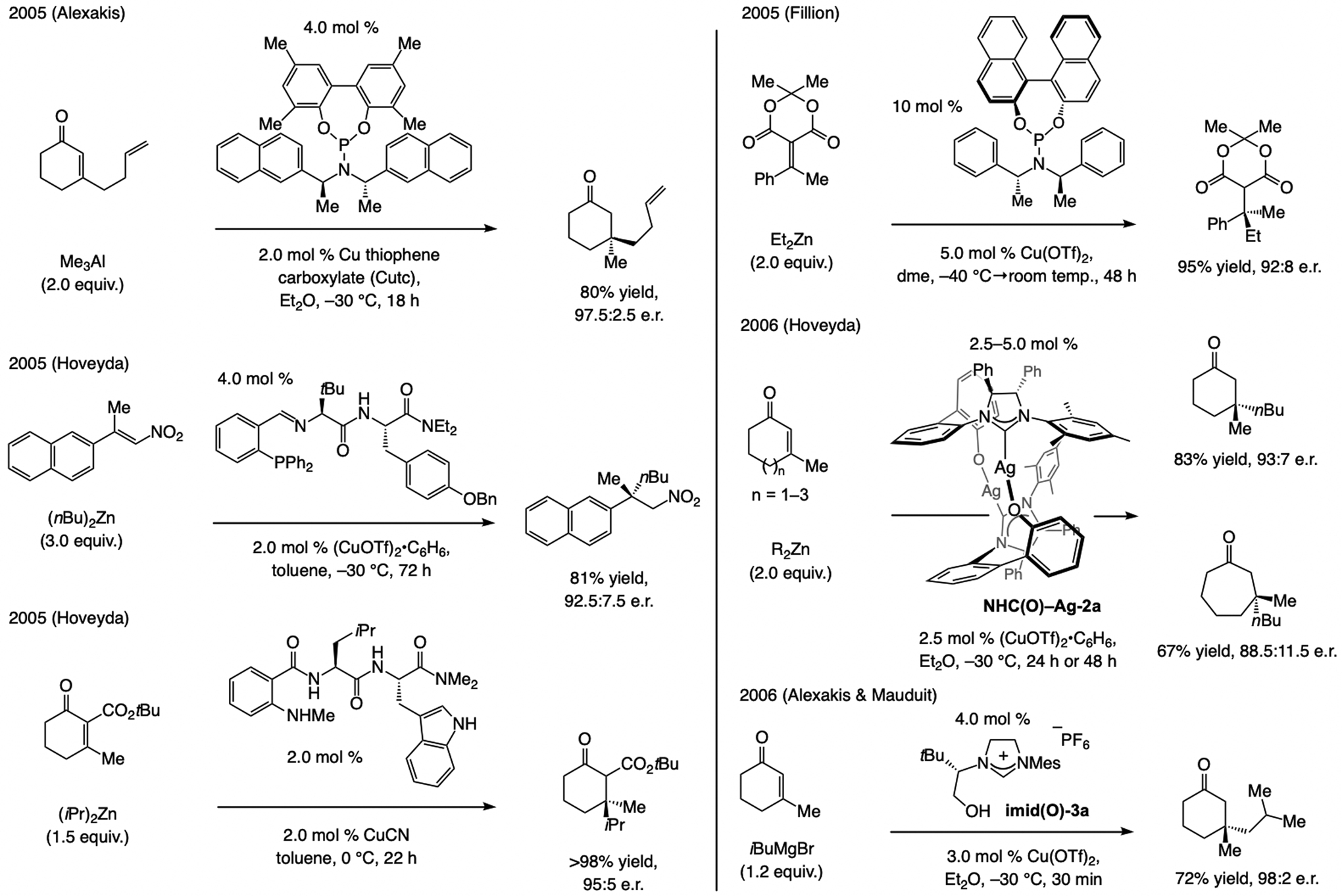
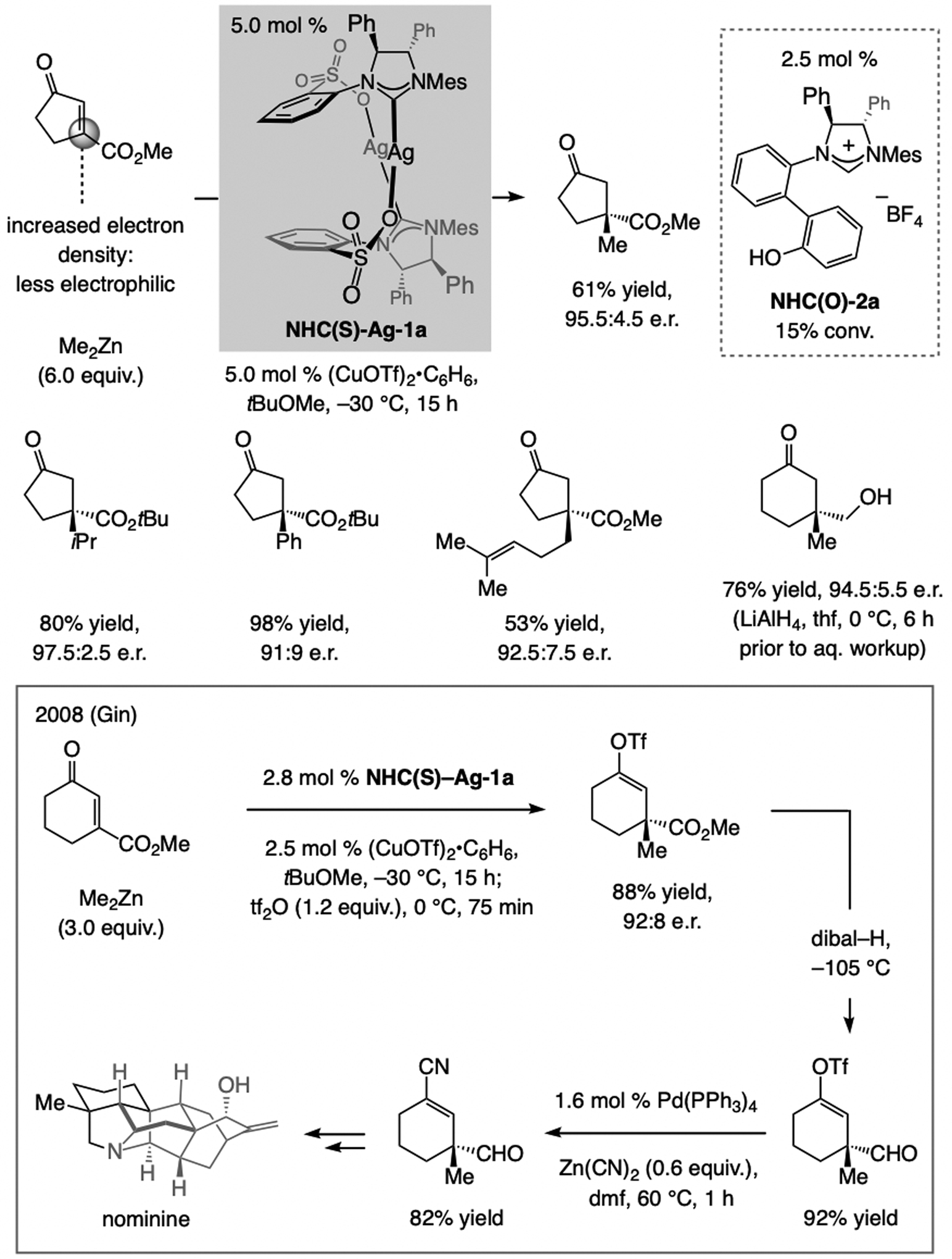
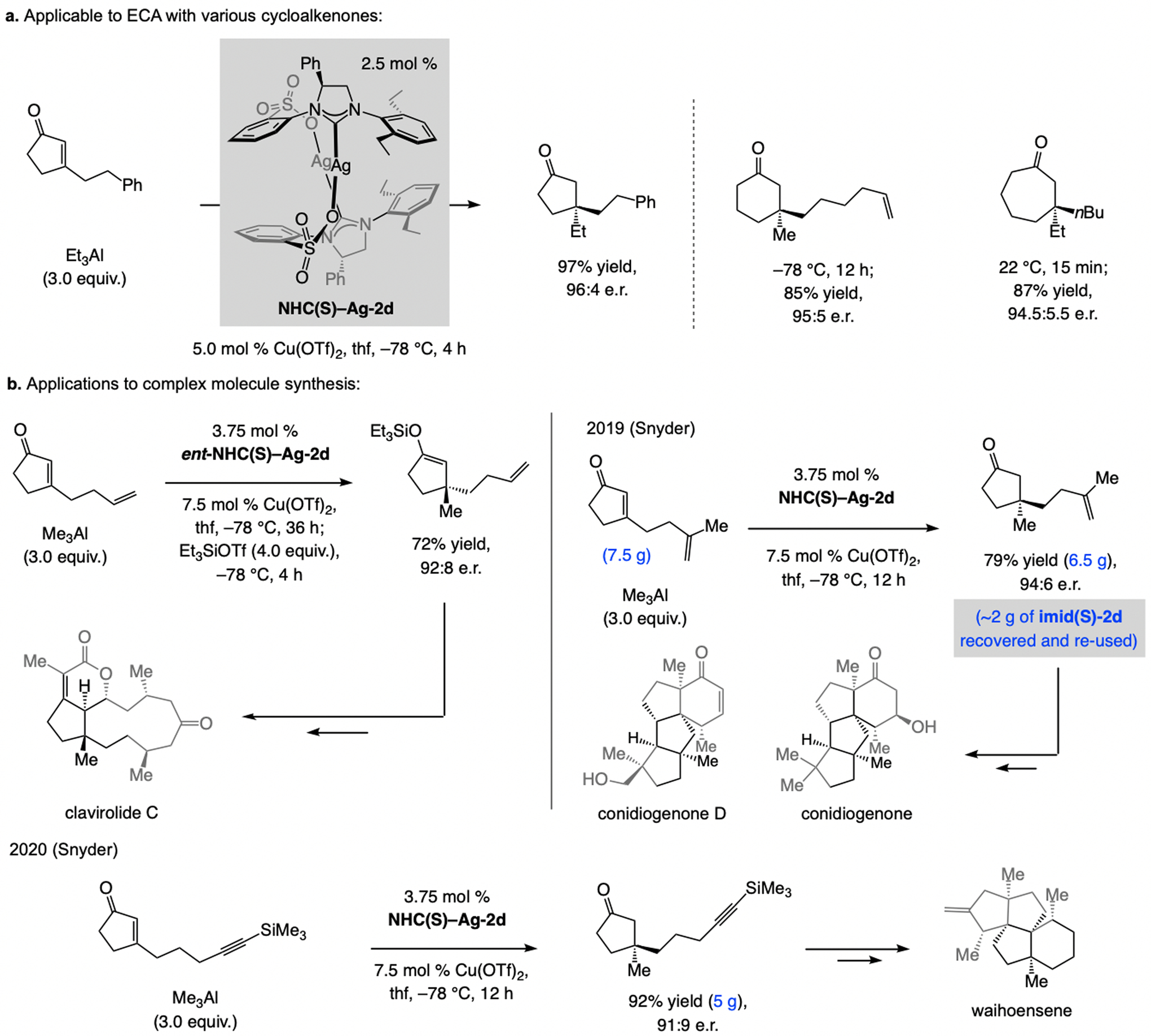
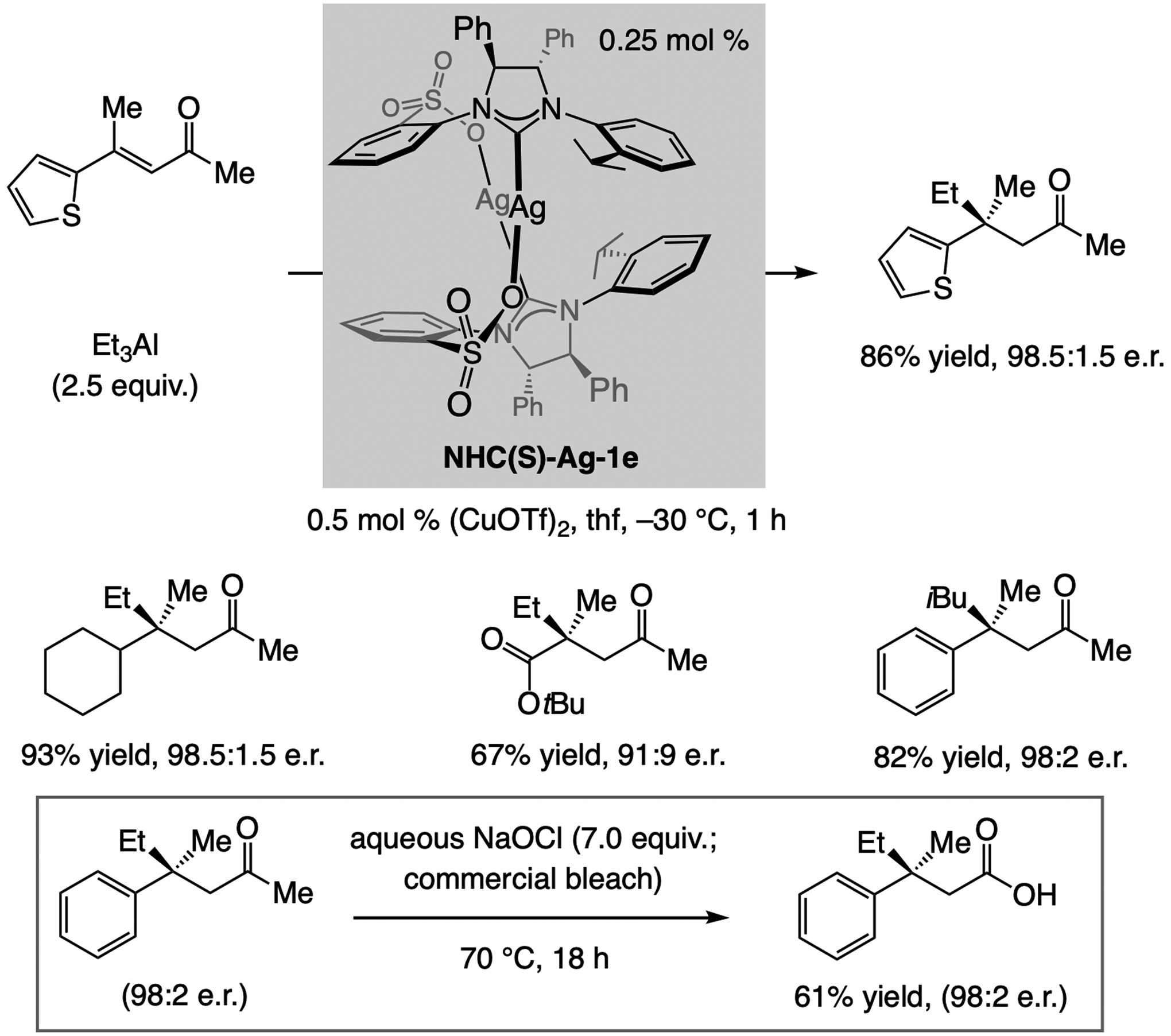
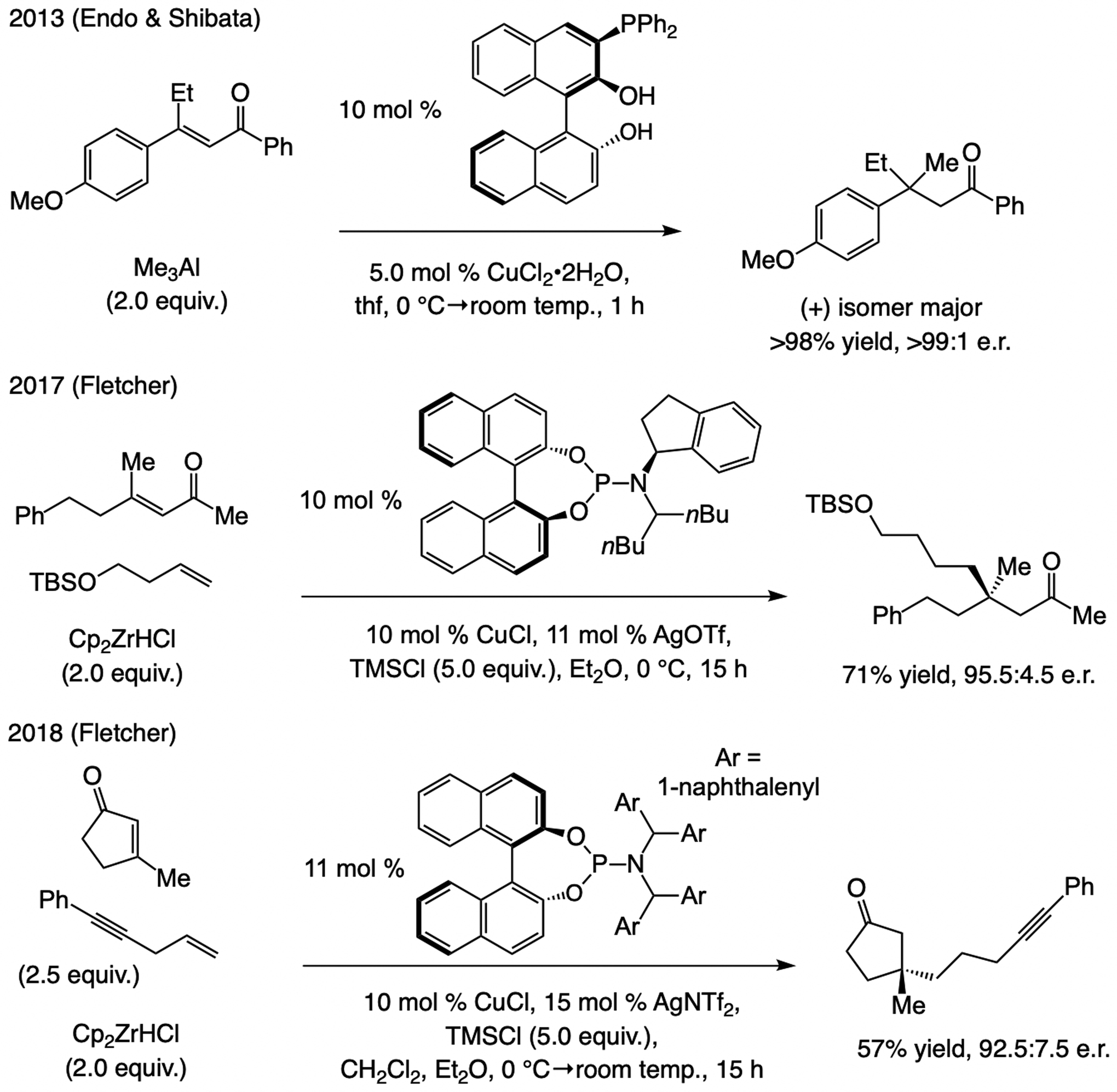
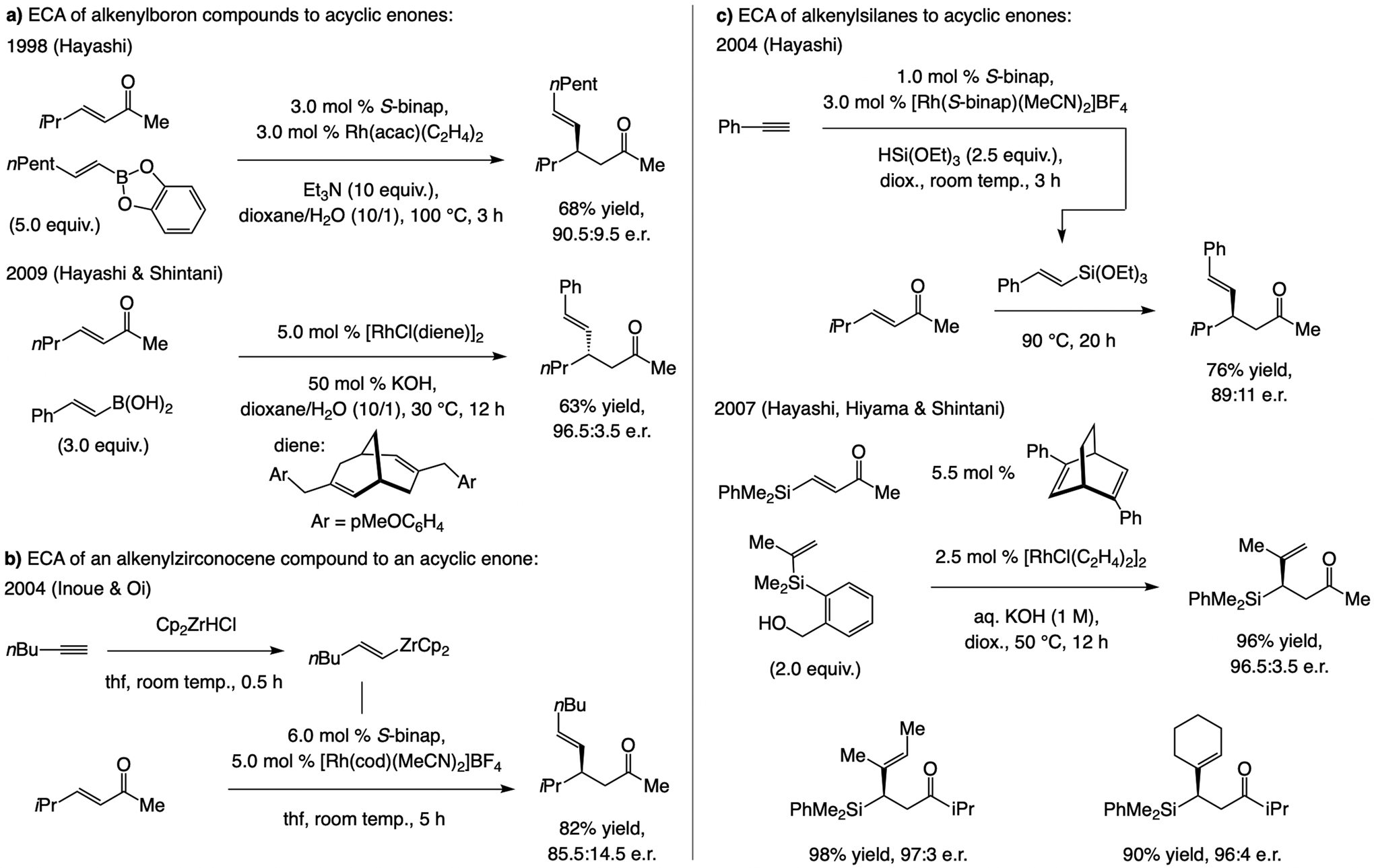
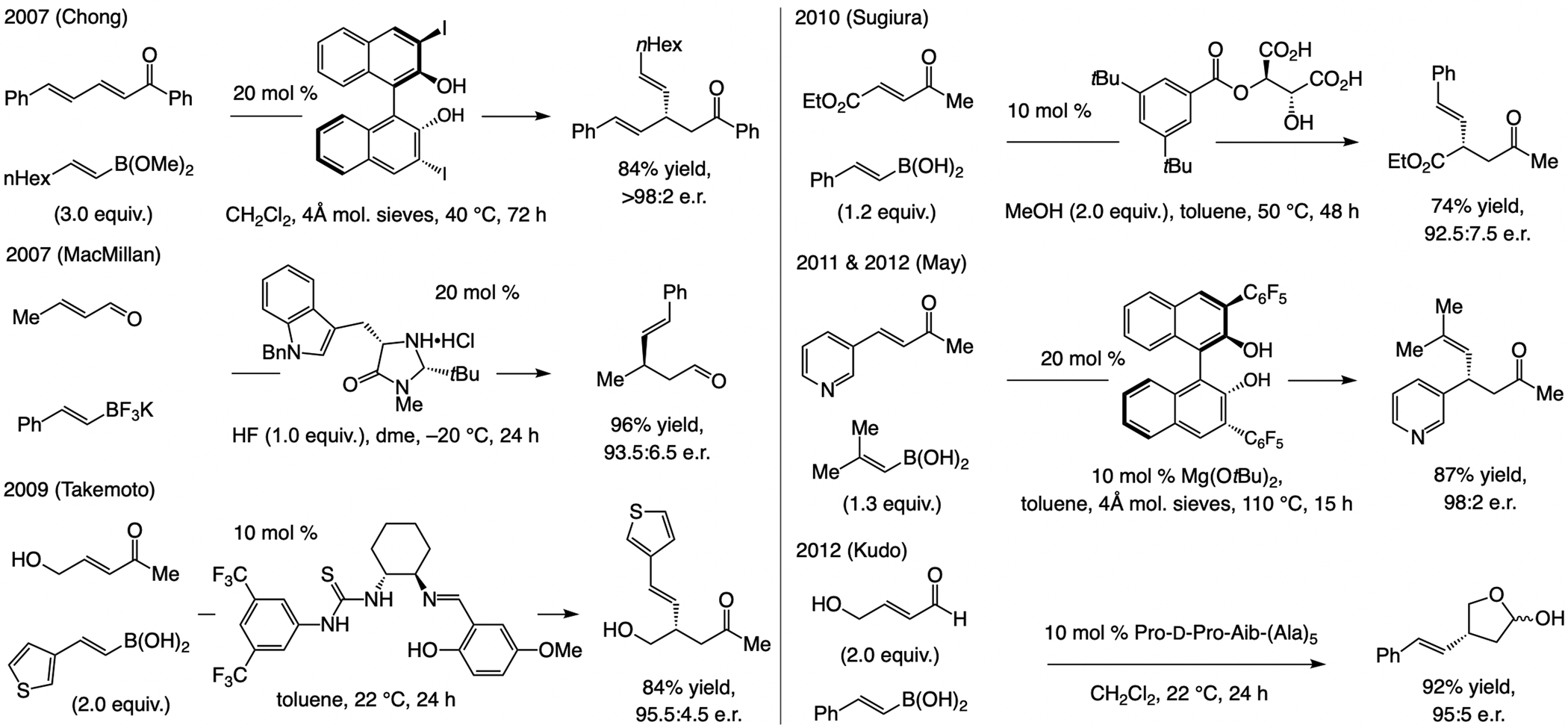
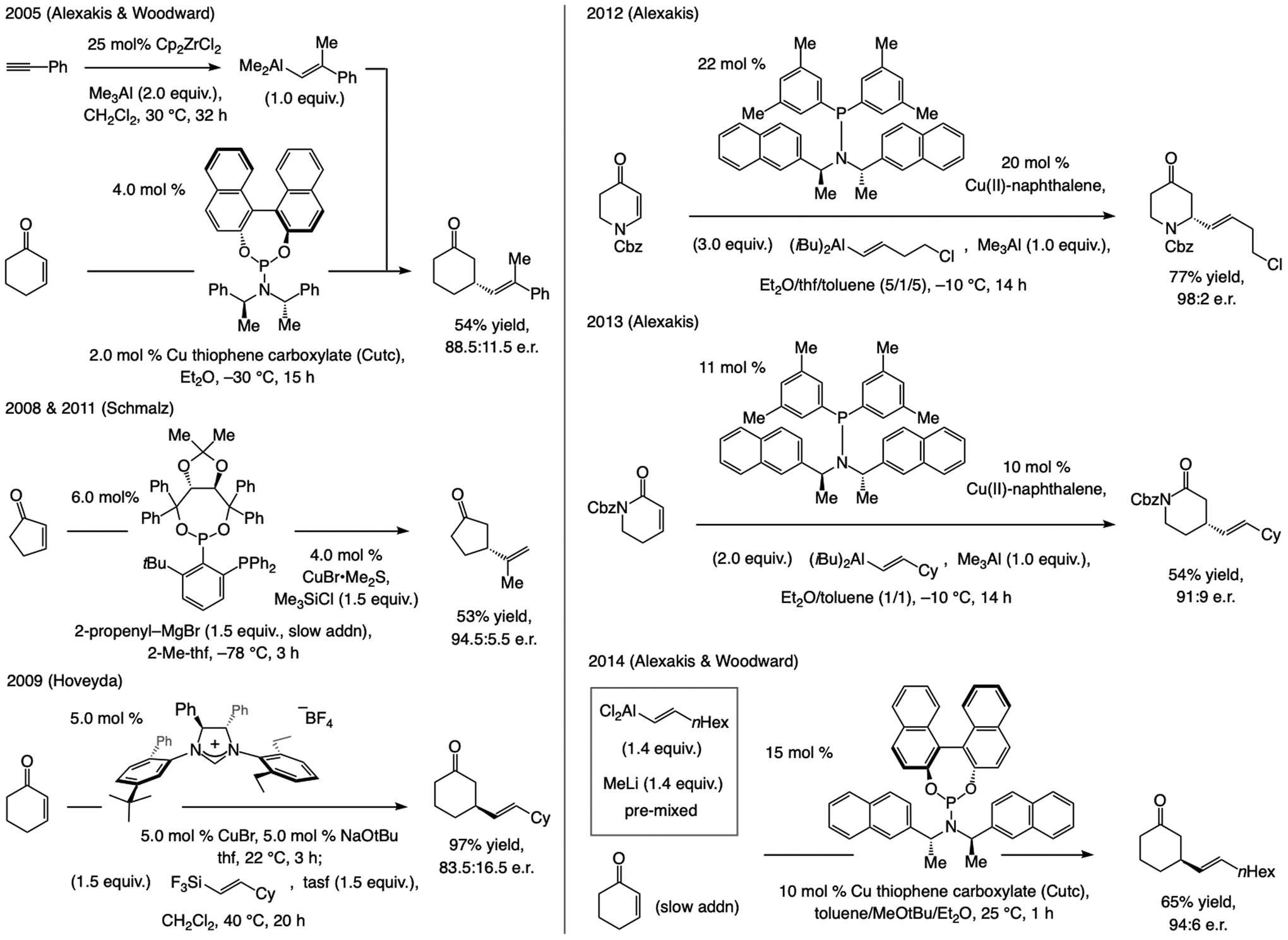
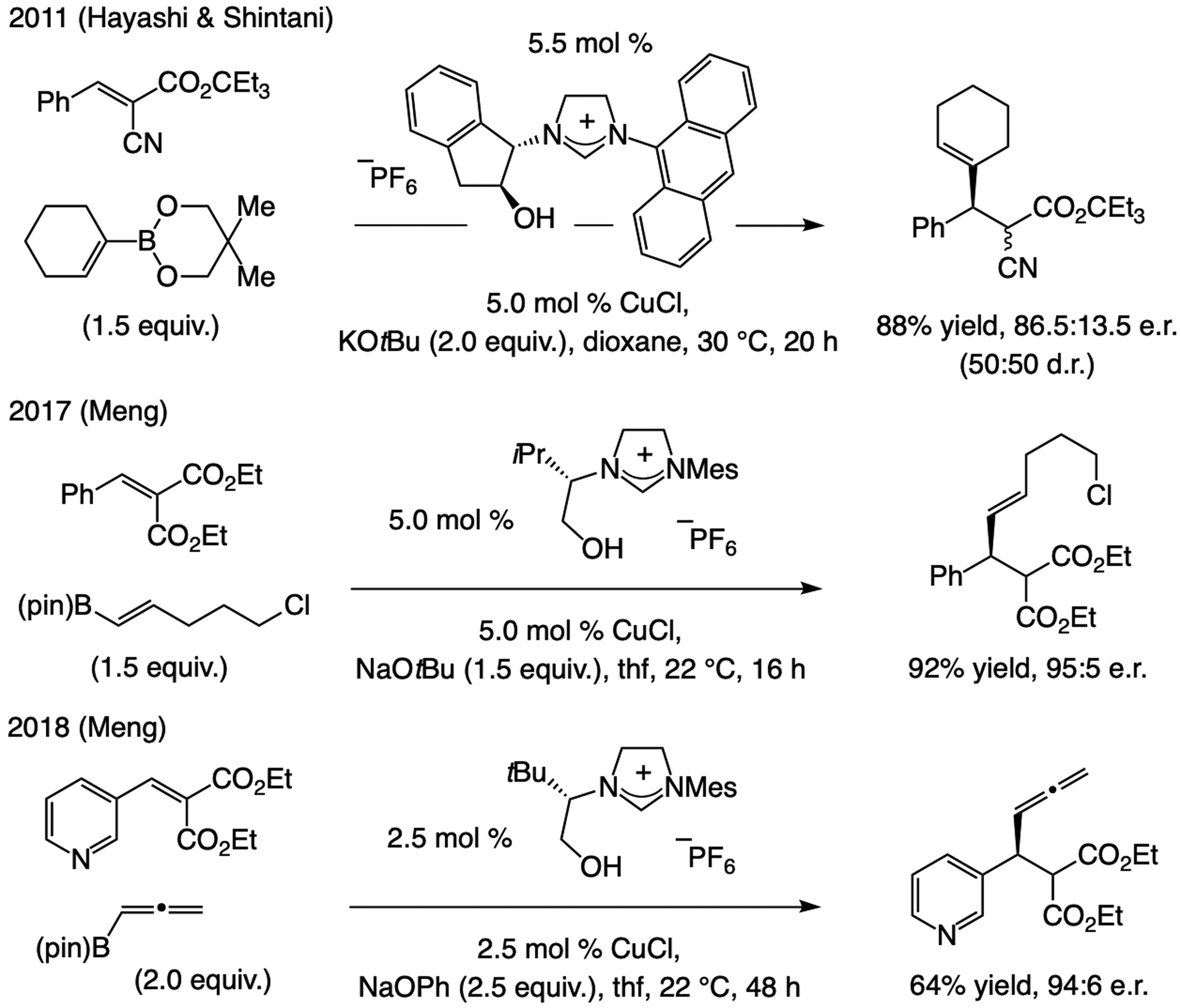
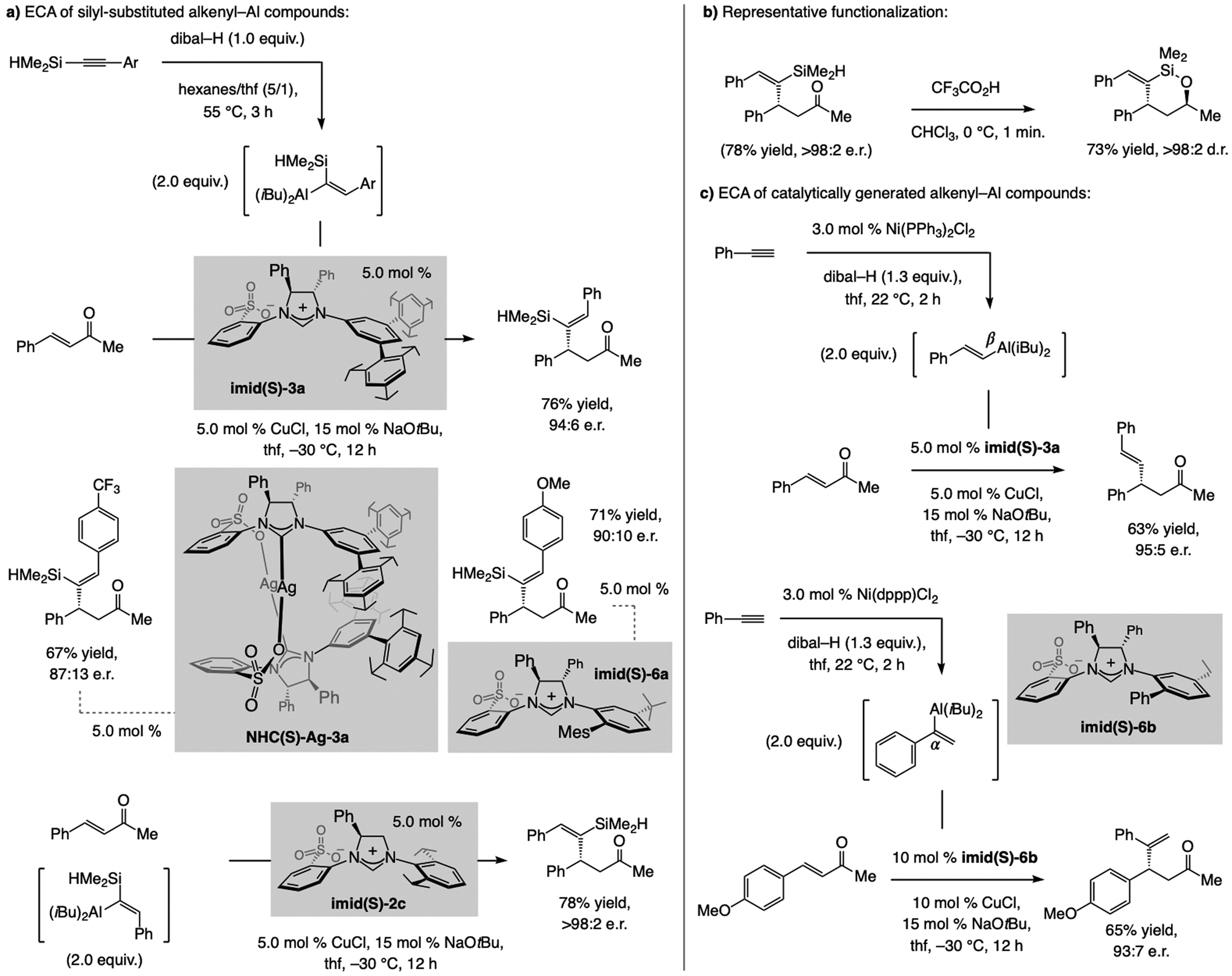
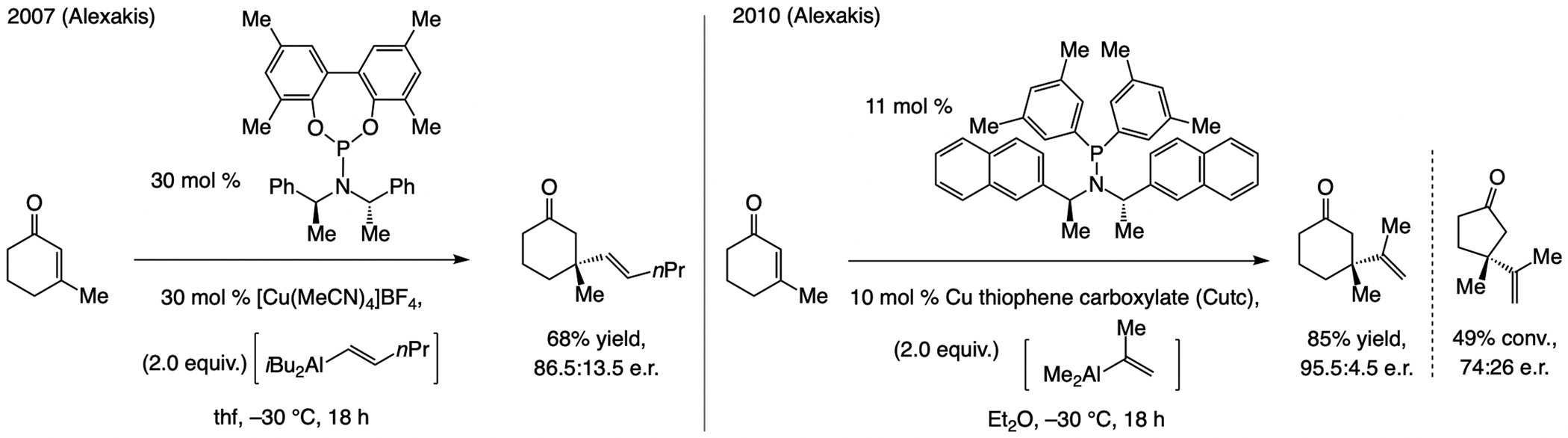
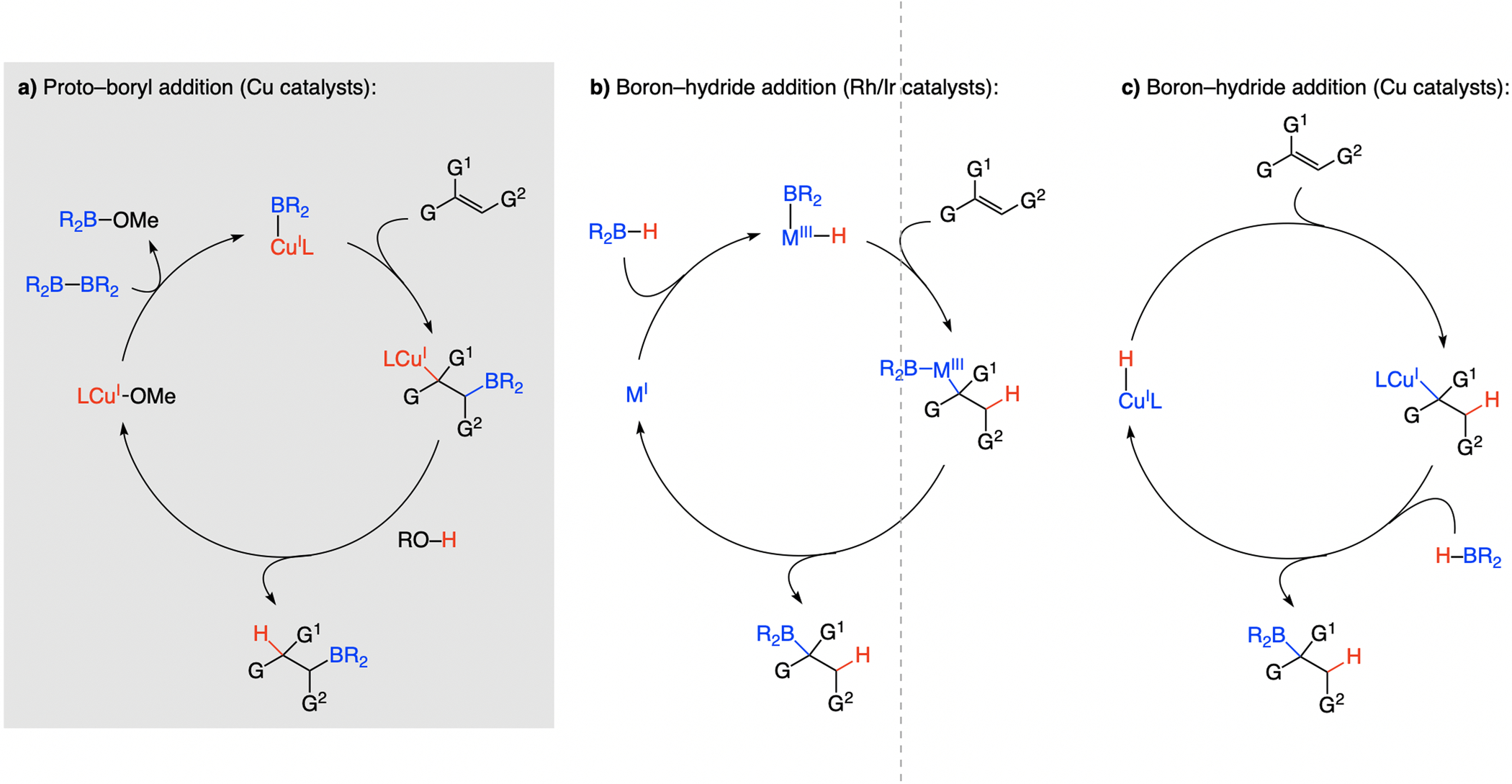
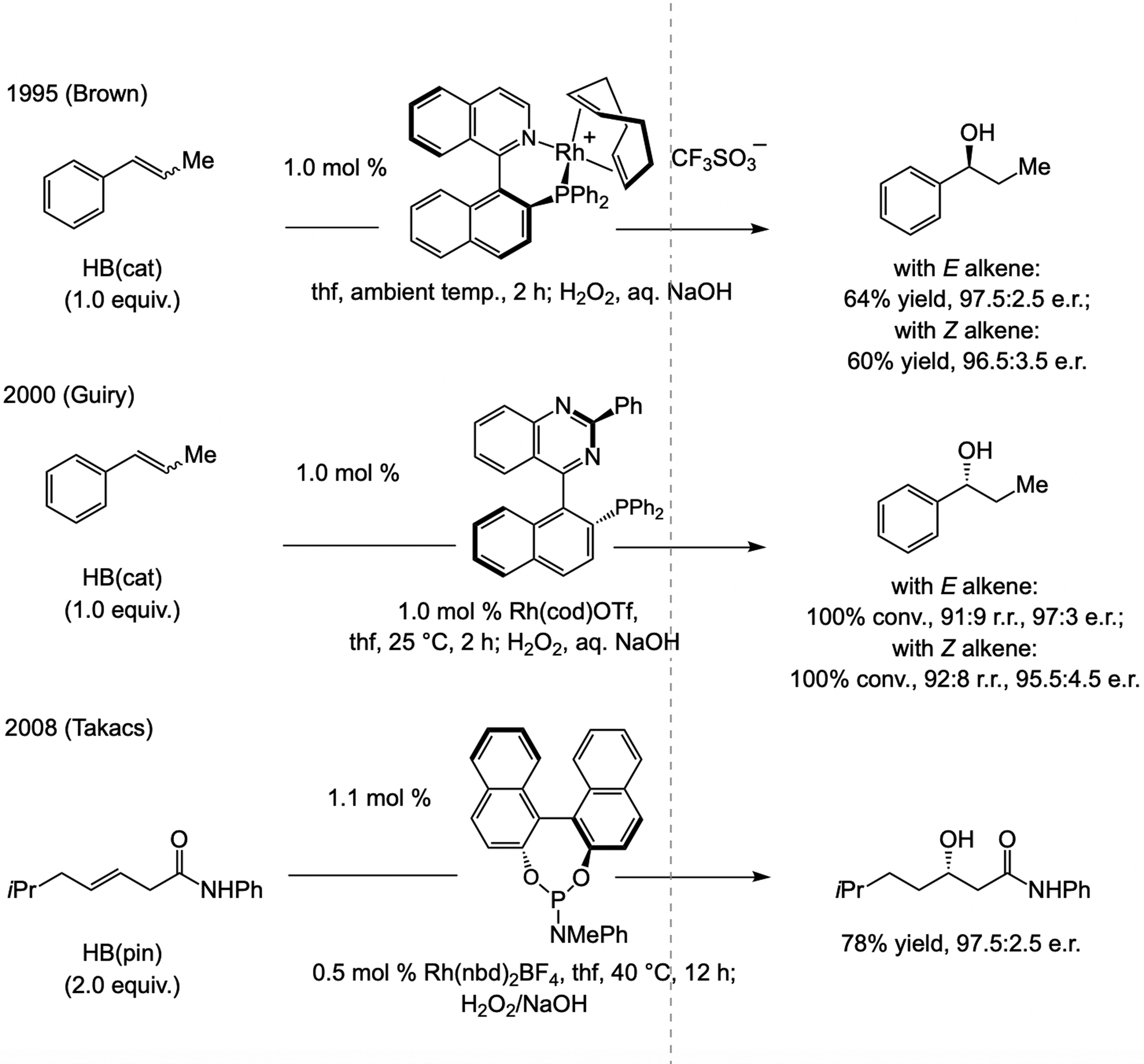
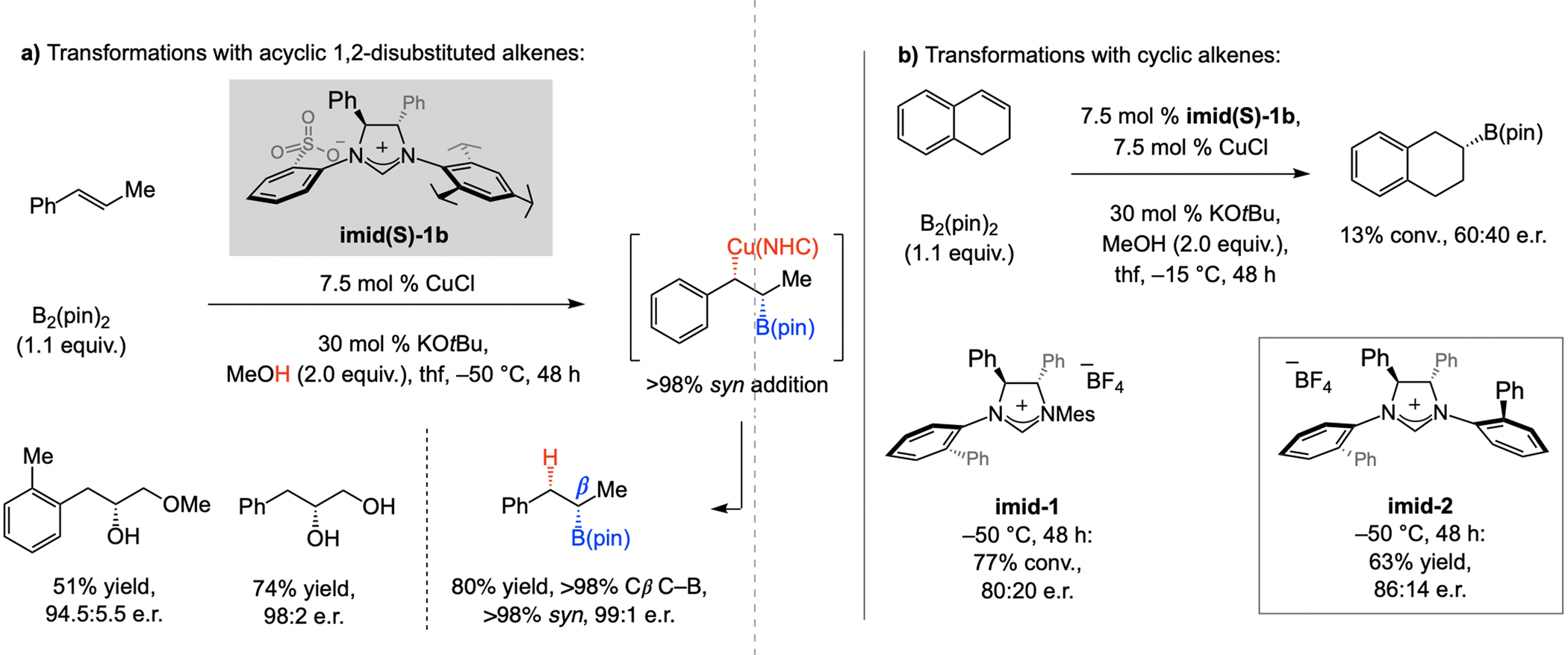
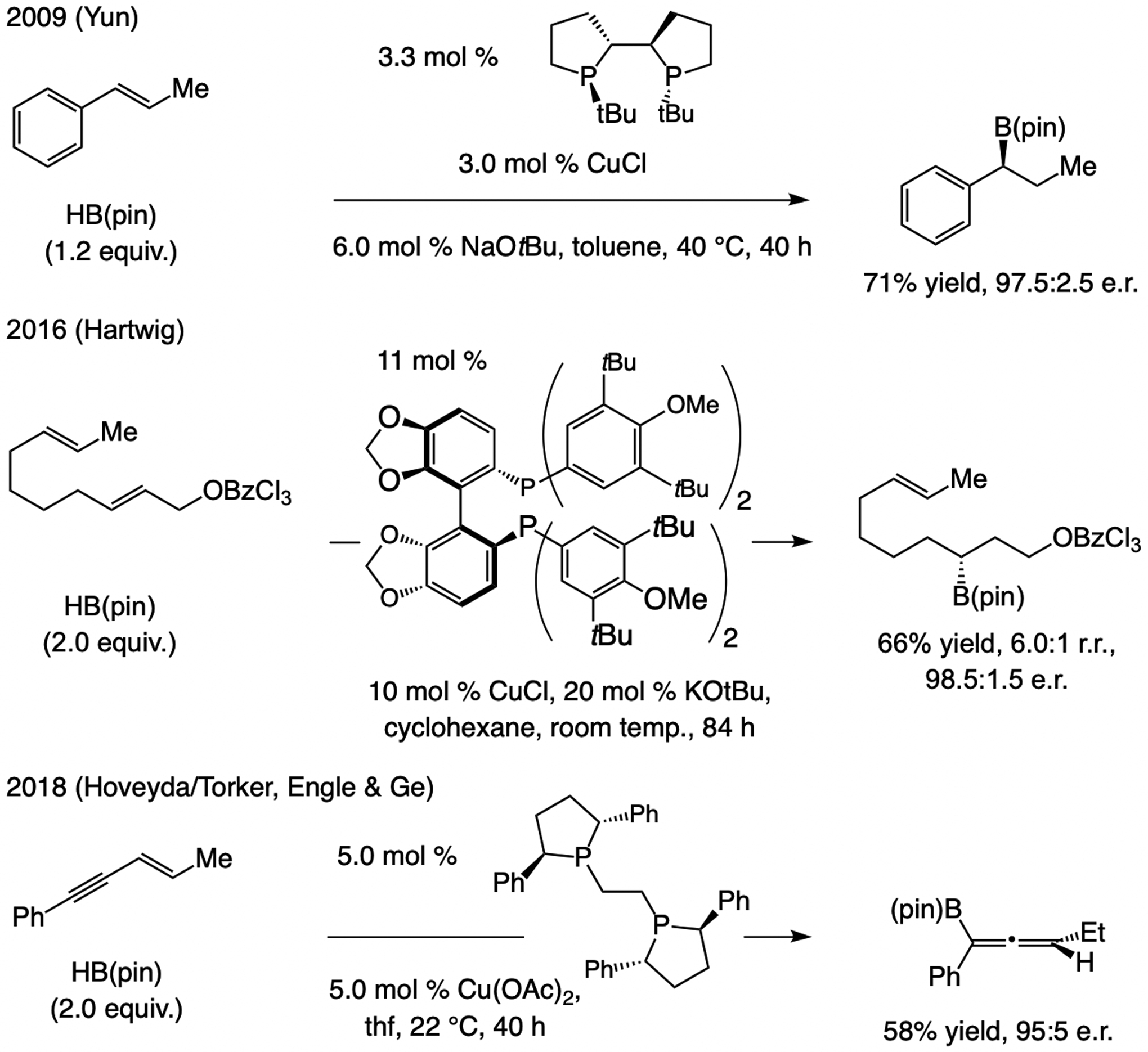
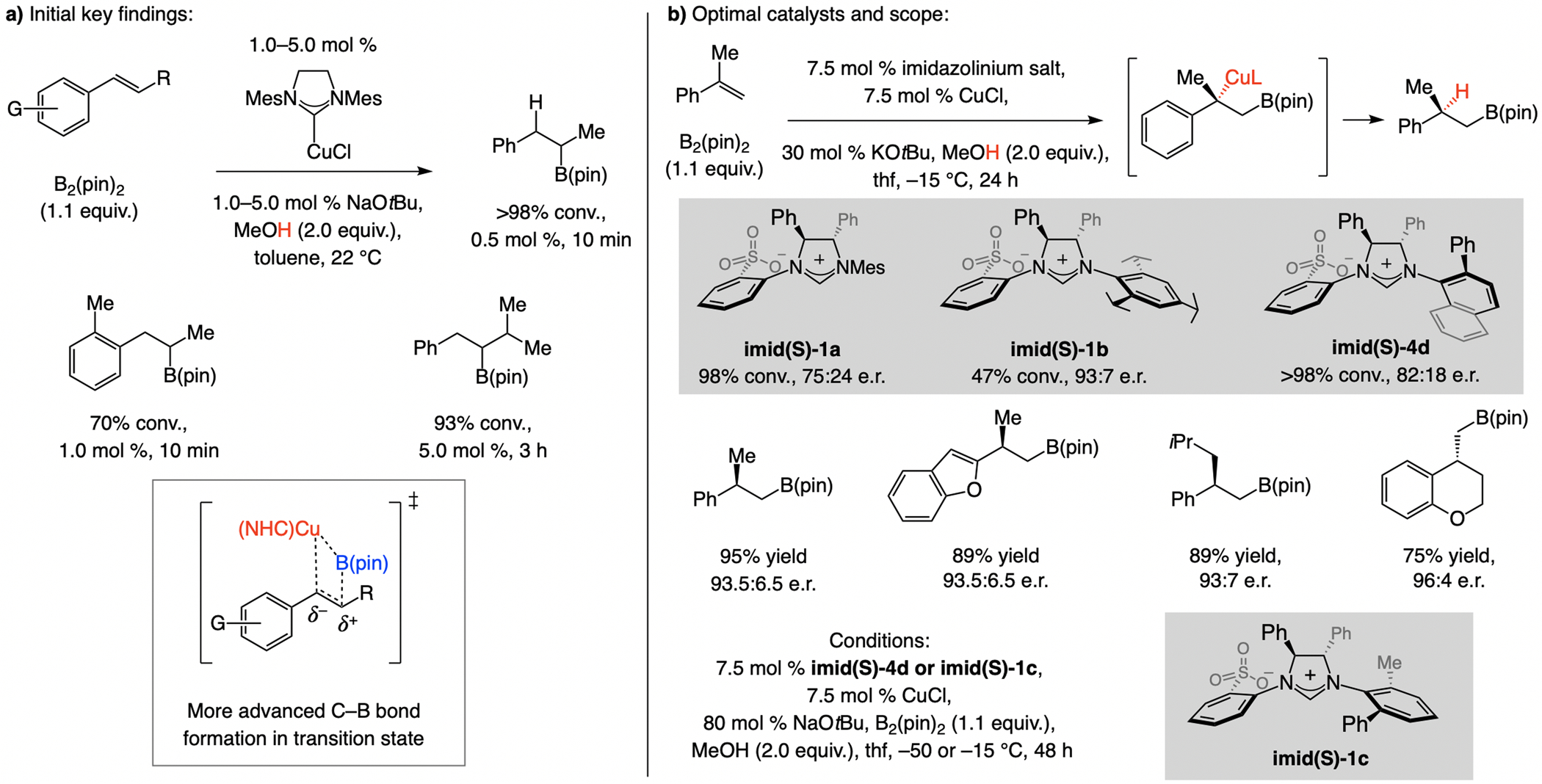
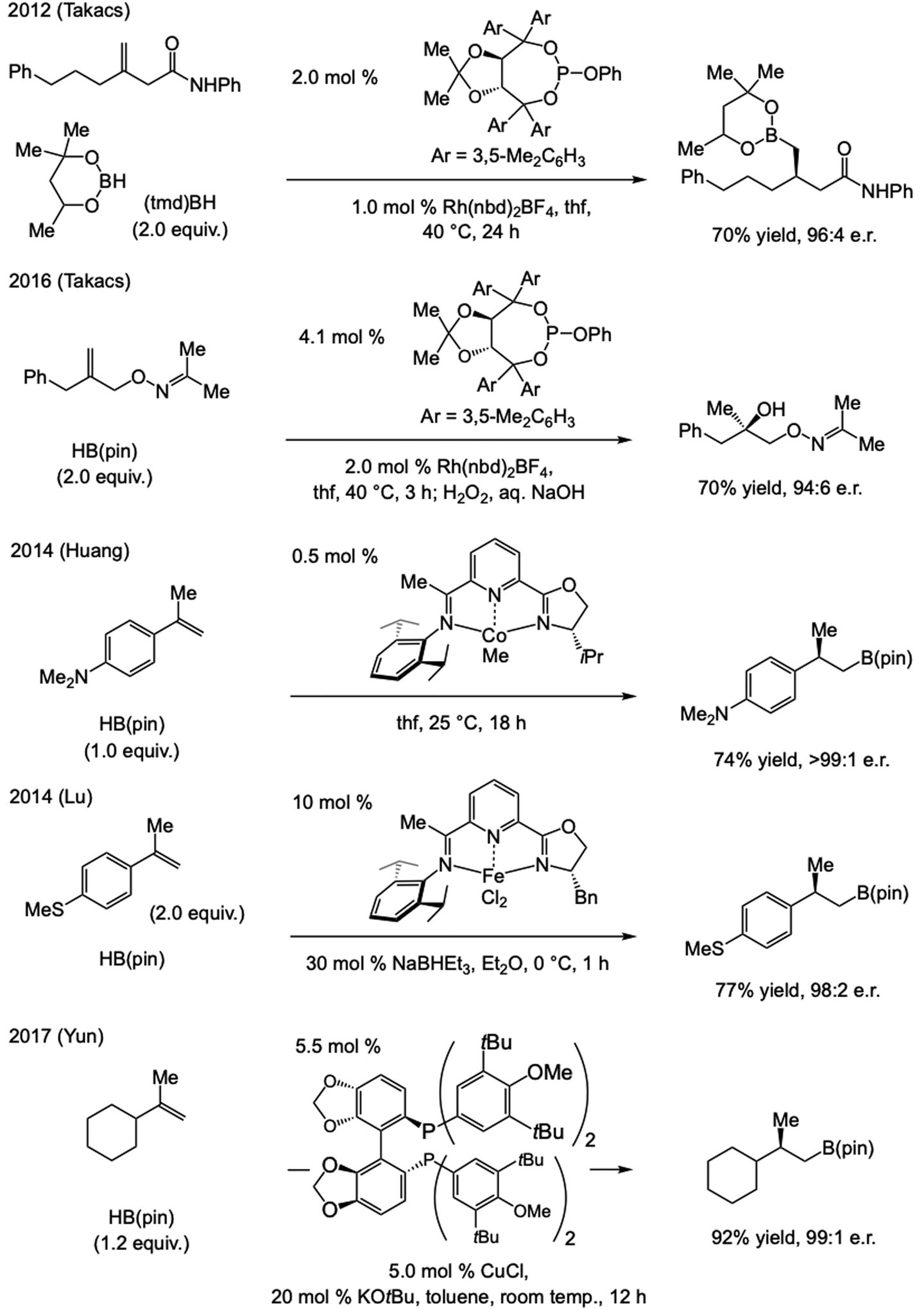
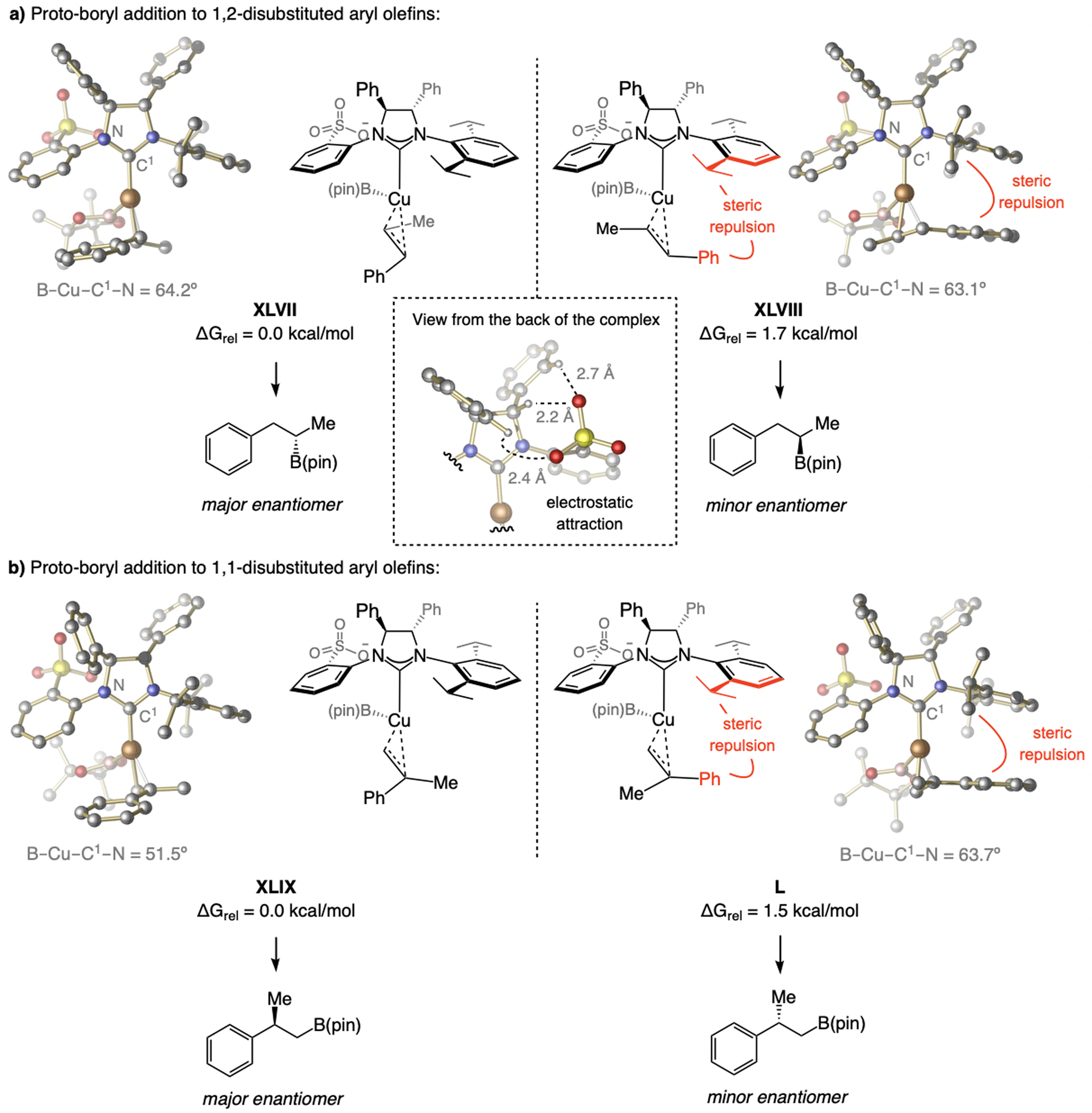
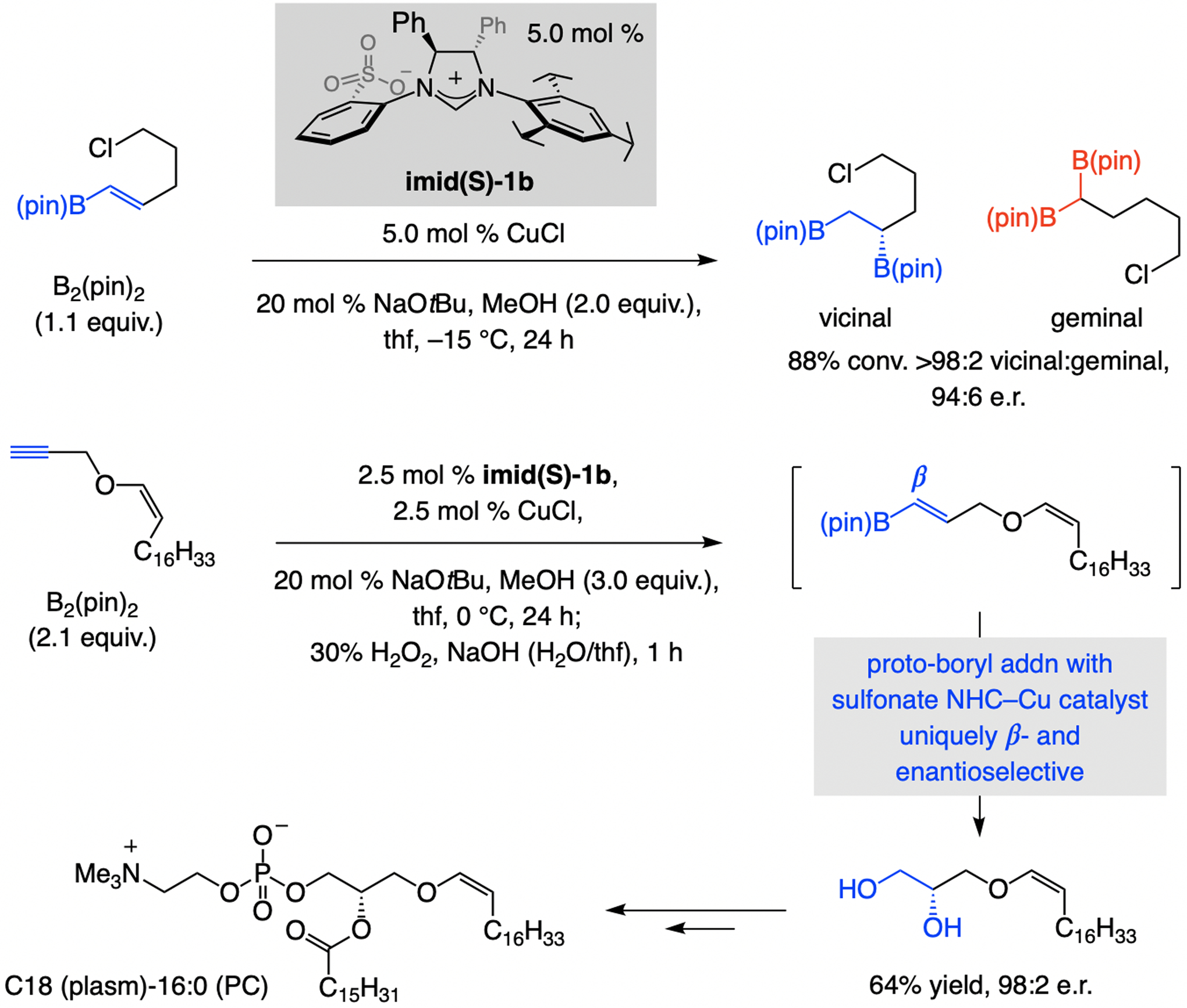
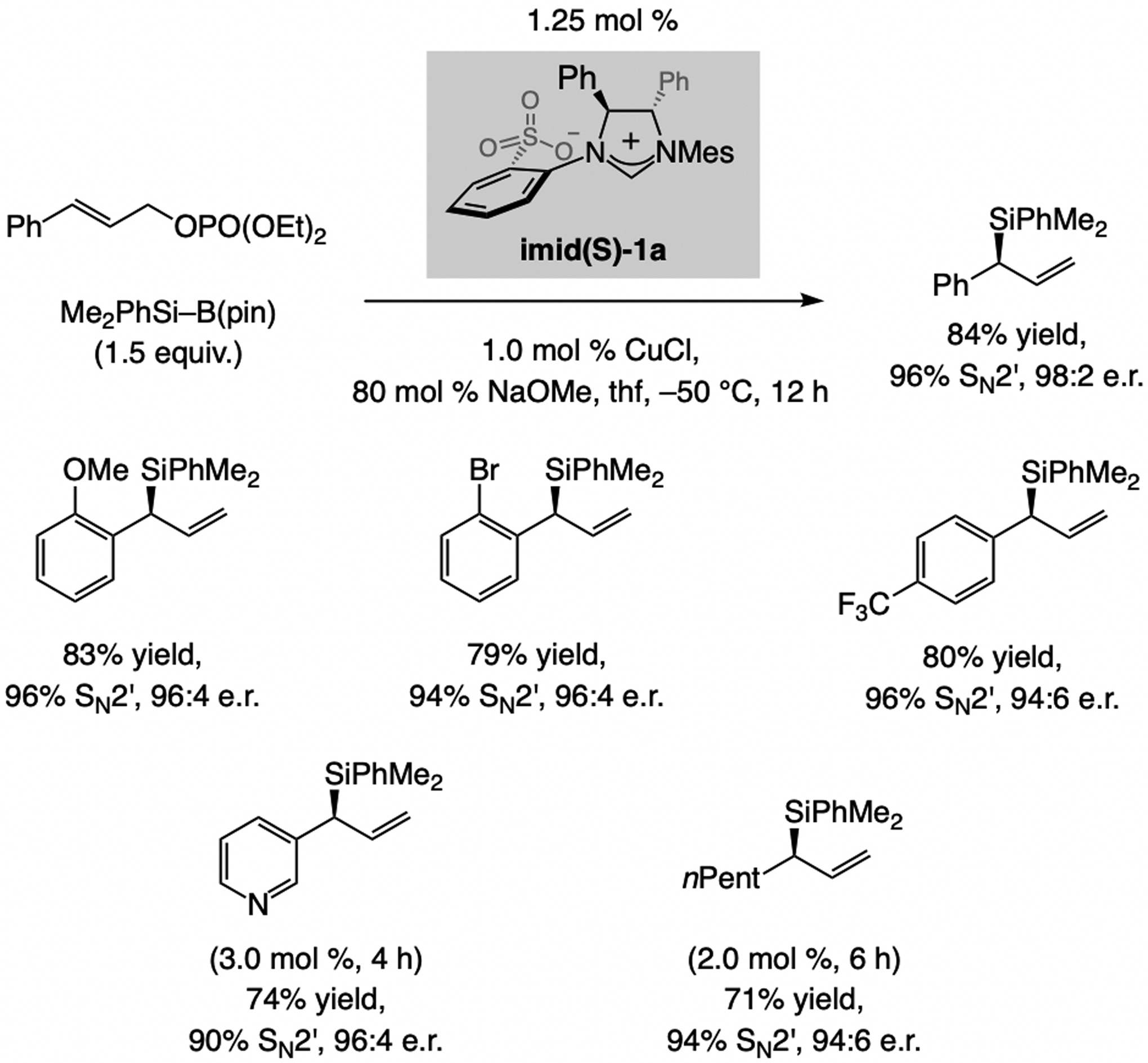
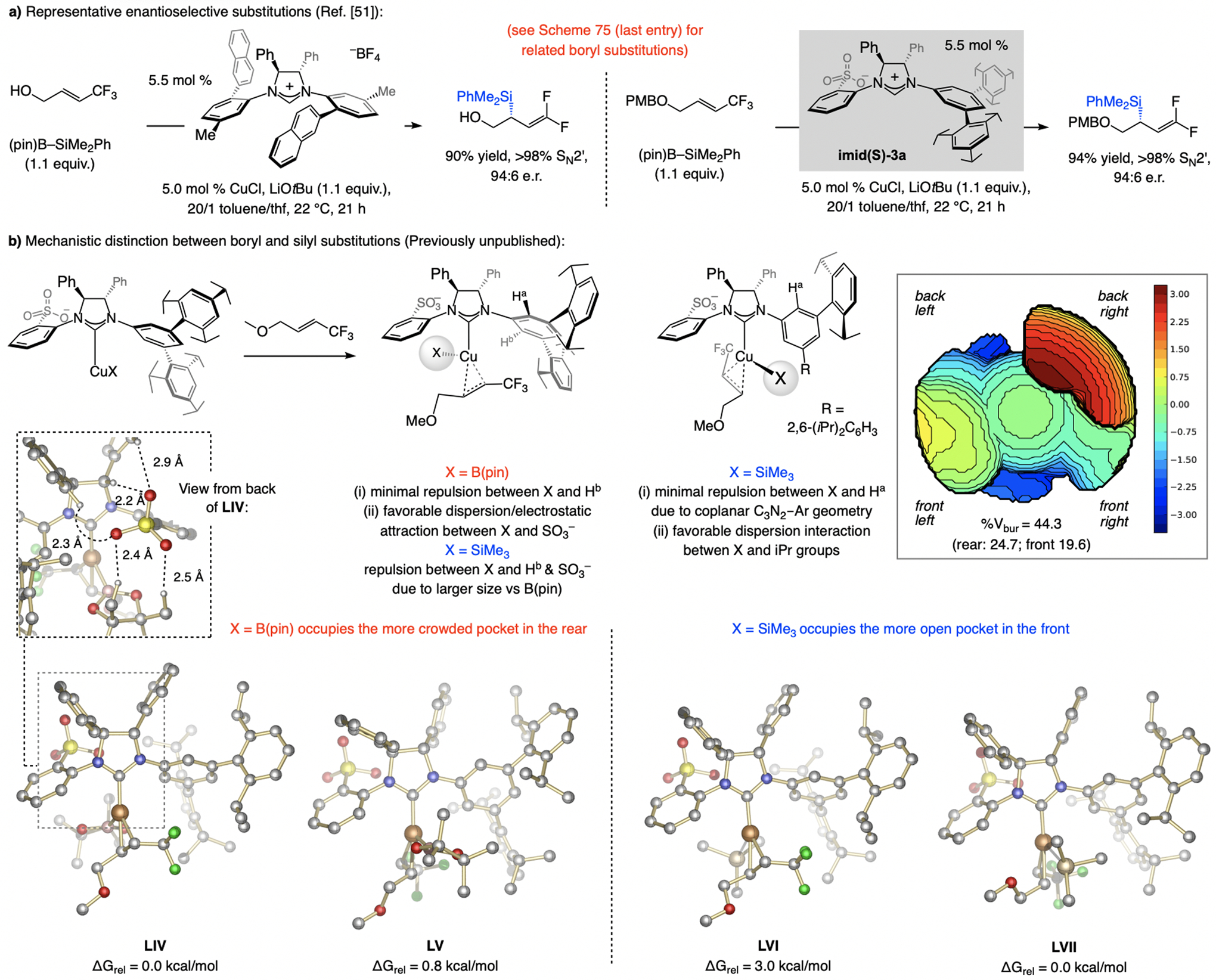
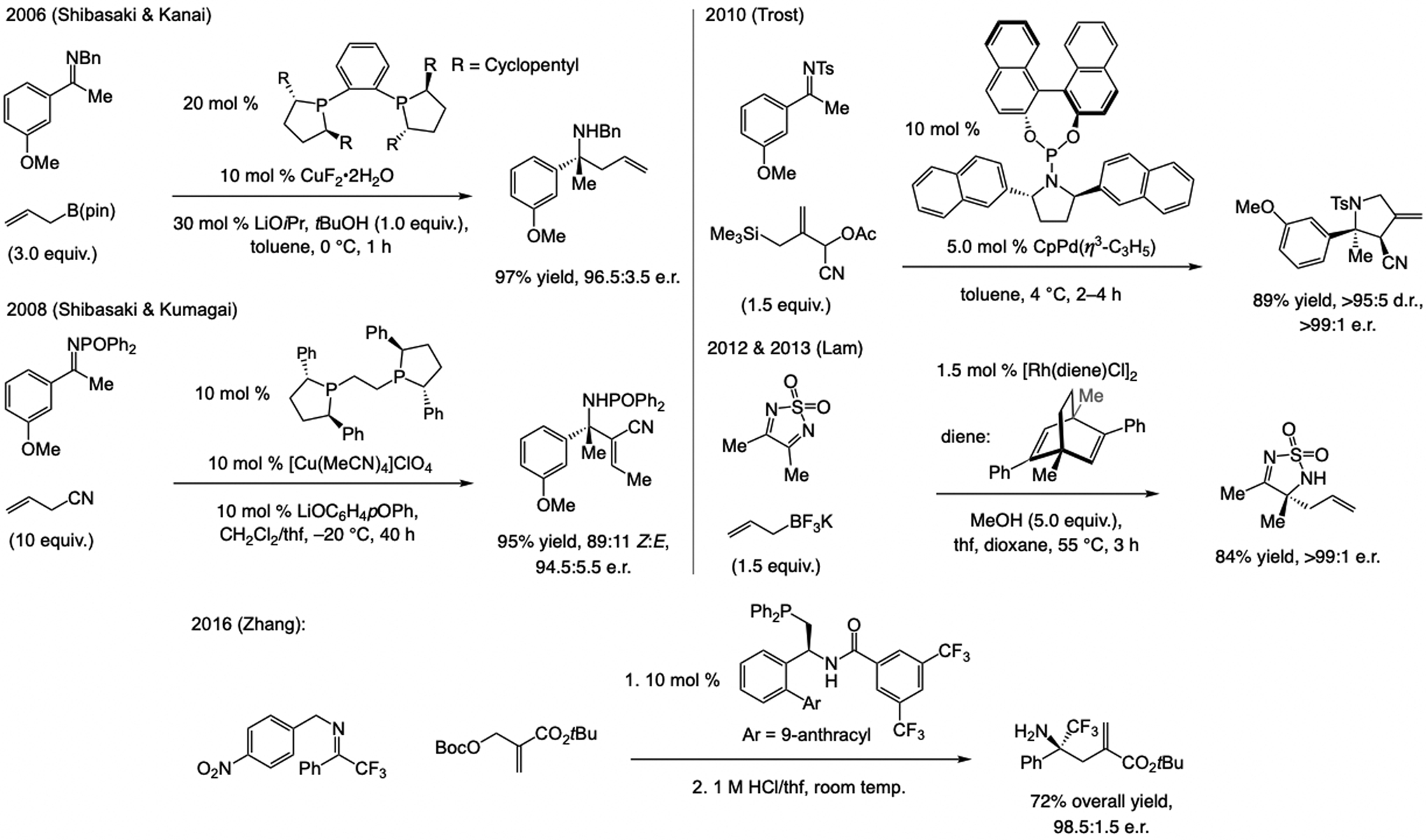
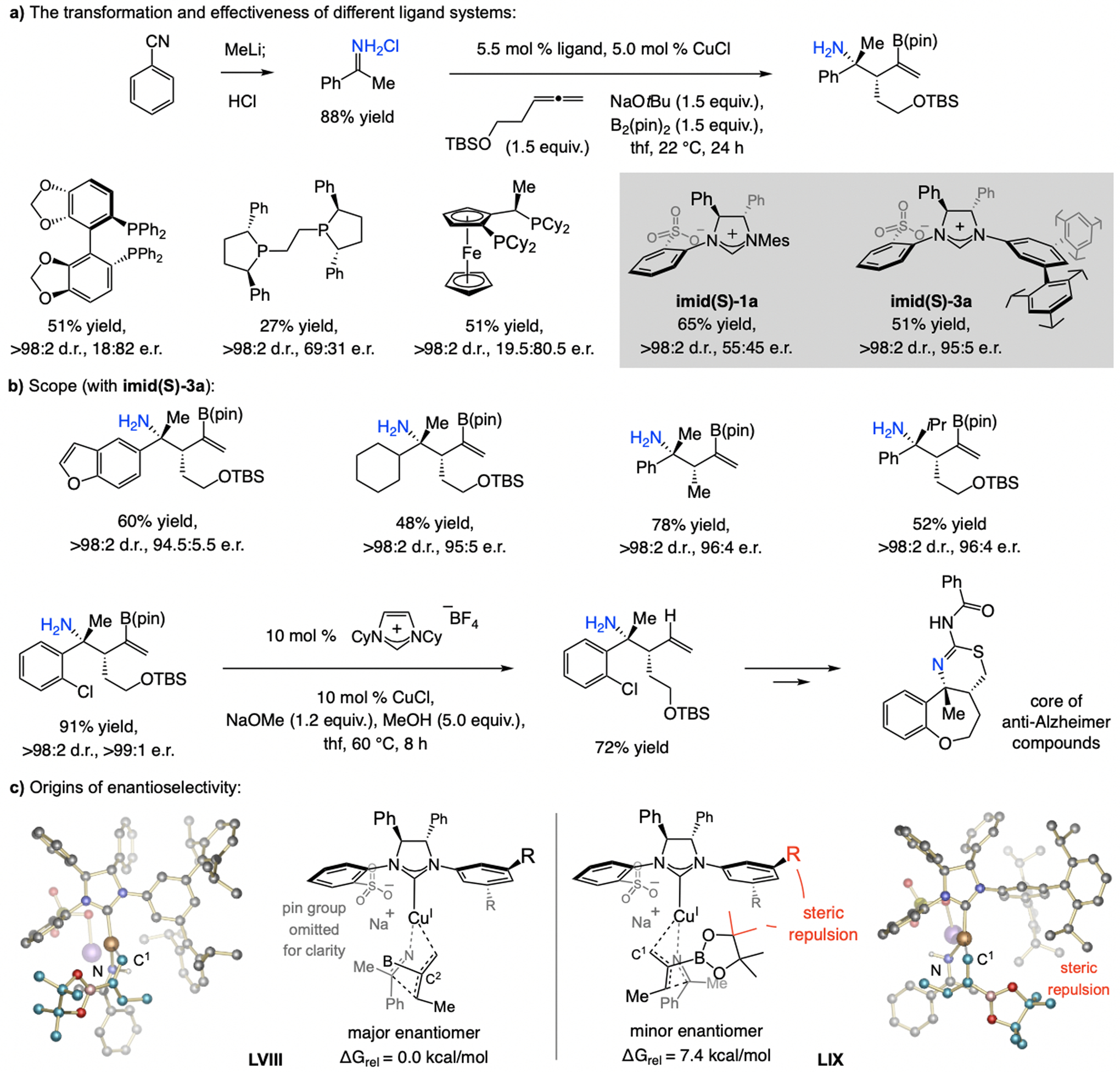
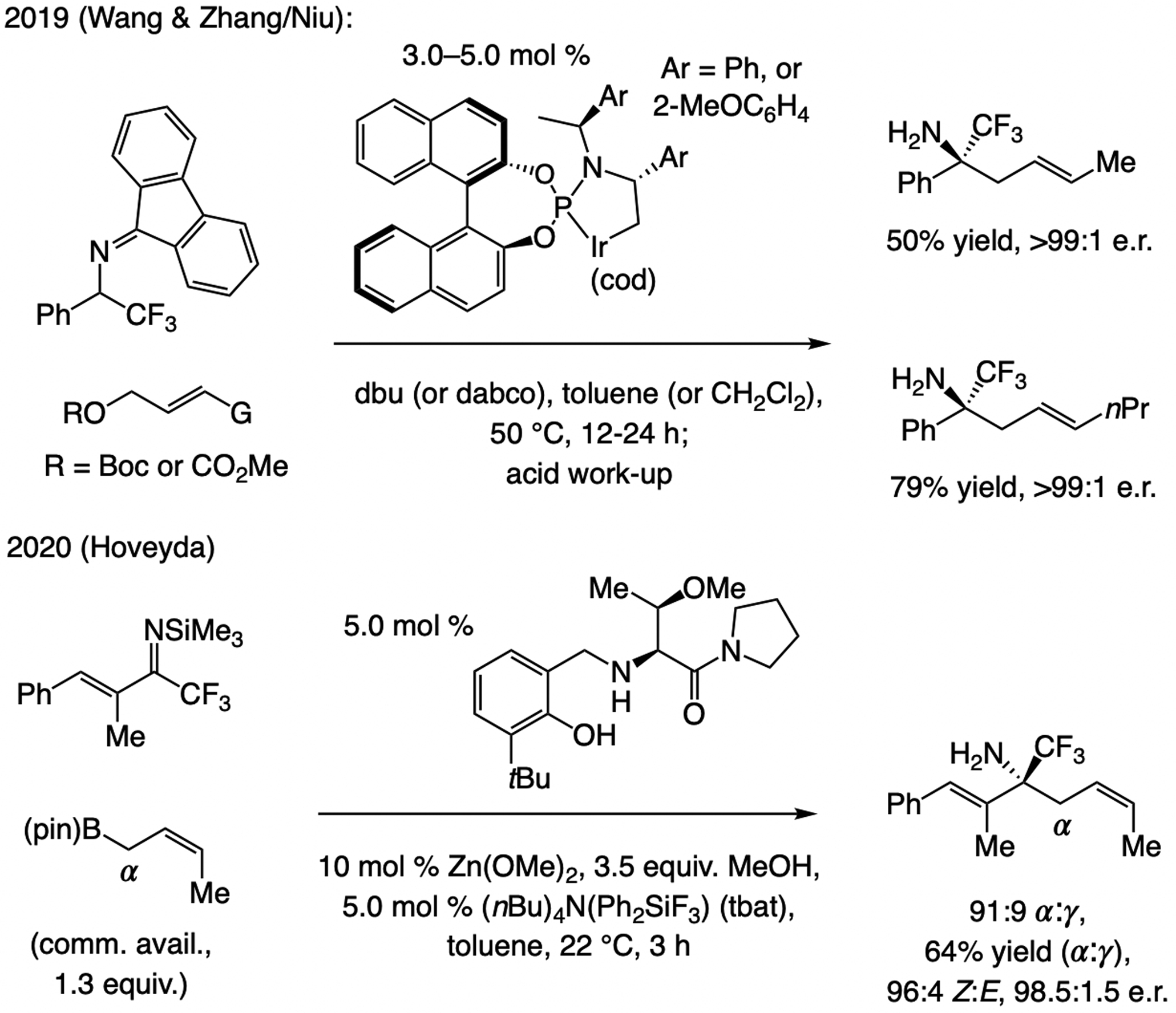
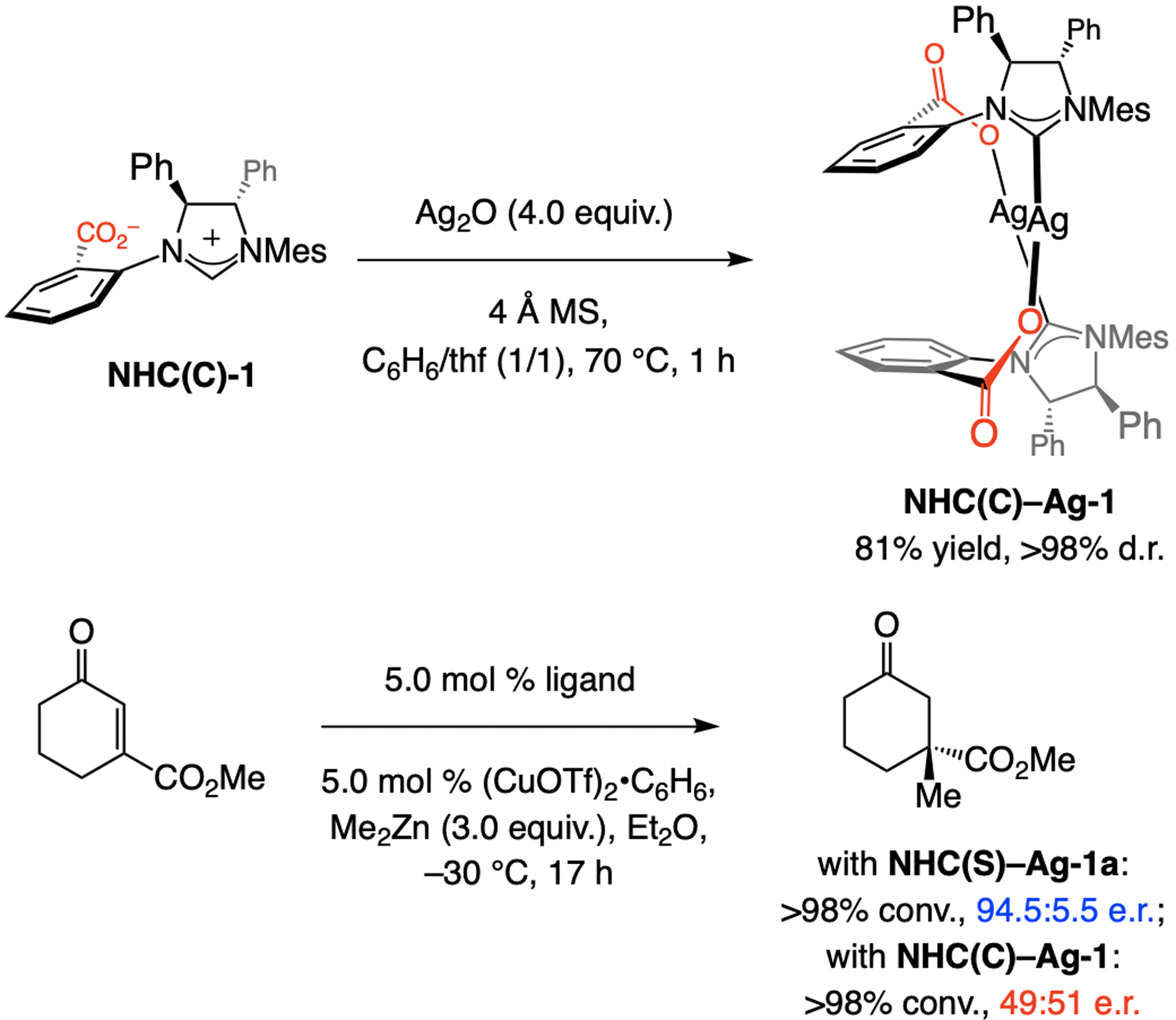
A copper-based complex that contains a sulfonate N-heterocyclic carbene ligand was first reported 15 years ago. Since then, these organometallic entities have proven to be uniquely effective in catalyzing an assortment of enantioselective transformations, including allylic substitutions, conjugate additions, proto-boryl additions to alkenes, boryl and silyl substitutions, hydride-allyl additions to alkenyl boronates, and additions of boron-containing allyl moieties to N-H ketimines. In this review article, we detail the shortcomings in the state-of-the-art that fueled the development of this air stable ligand class, members of which can be prepared on multigram scale. For each reaction type, when relevant, the prior art at the time of the advance involving sulfonate NHC-Cu catalysts and/or subsequent key developments are briefly analyzed, and the relevance of the advance to efficient and enantioselective total or formal synthesis of biologically active molecules is underscored. Mechanistic analysis of the structural attributes of sulfonate NHC-Cu catalysts that are responsible for their ability to facilitate transformations with high efficiency as well as regio- and enantioselectivity are detailed. This review contains several formerly undisclosed methodological advances and mechanistic analyses, the latter of which constitute a revision of previously reported proposals.
Keywords: NHC ligands; allylic substitutions; conjugate additions; copper; enantioselective catalysis.
© 2020 Wiley-VCH GmbH.
Figures
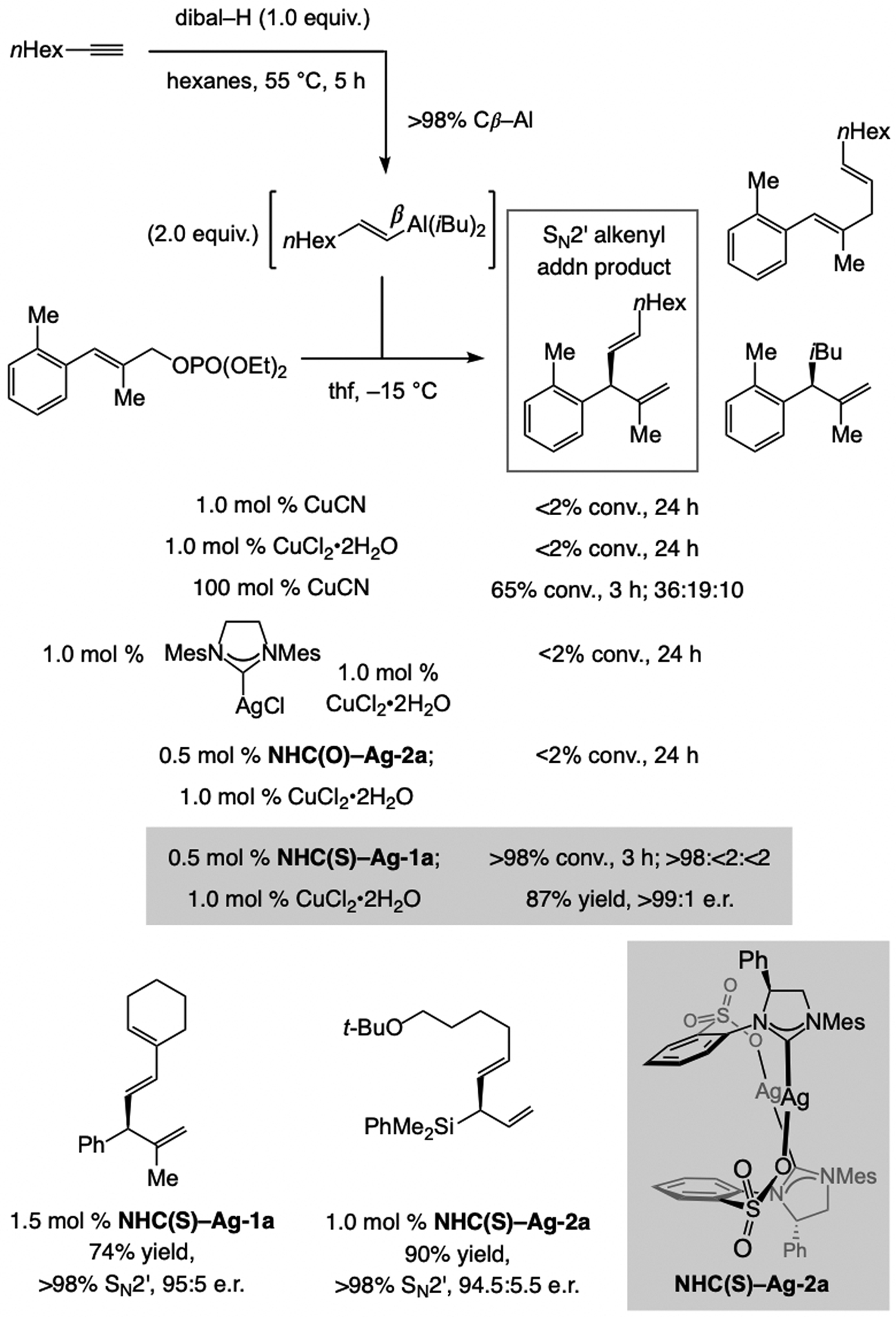
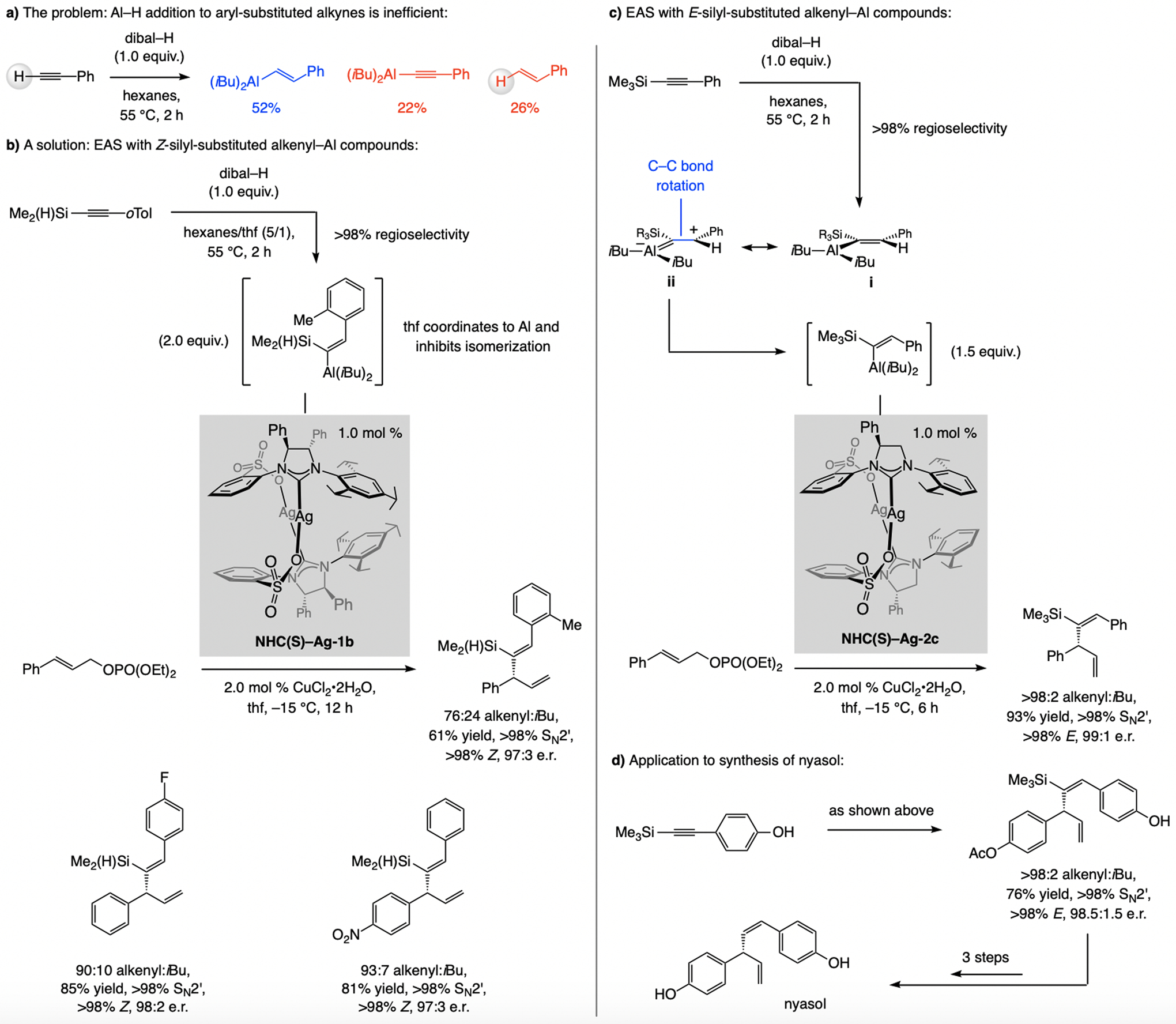
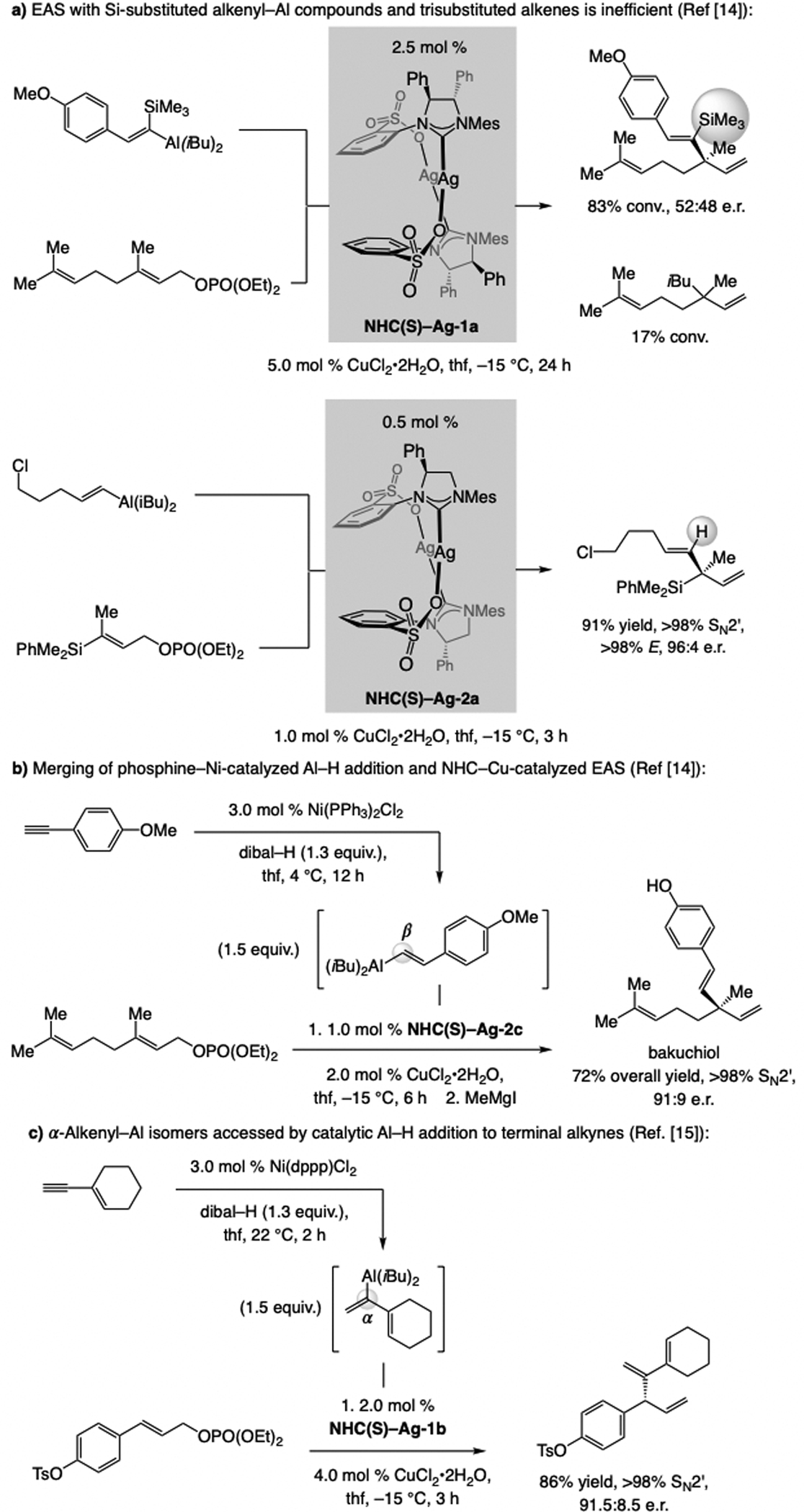
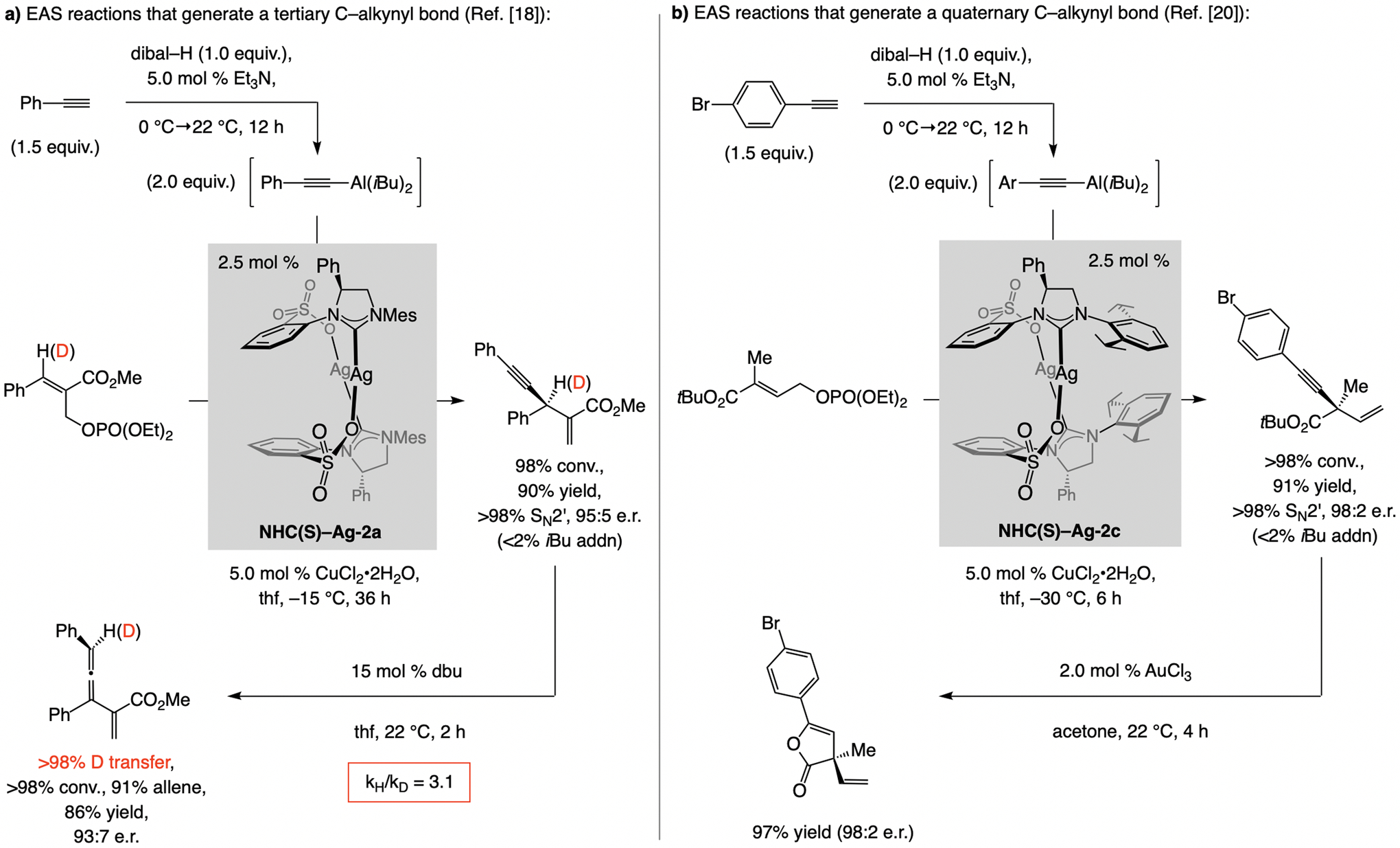
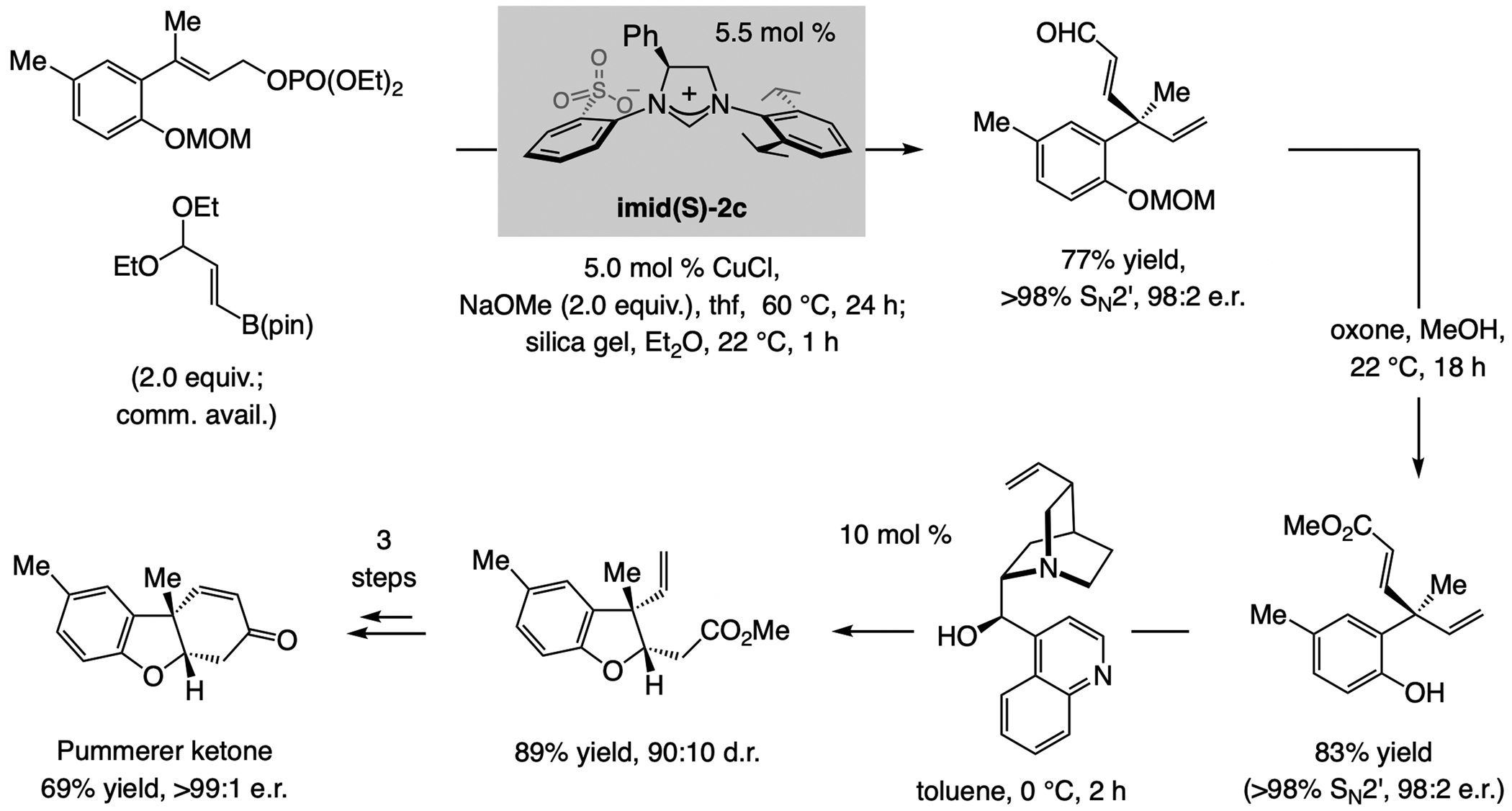
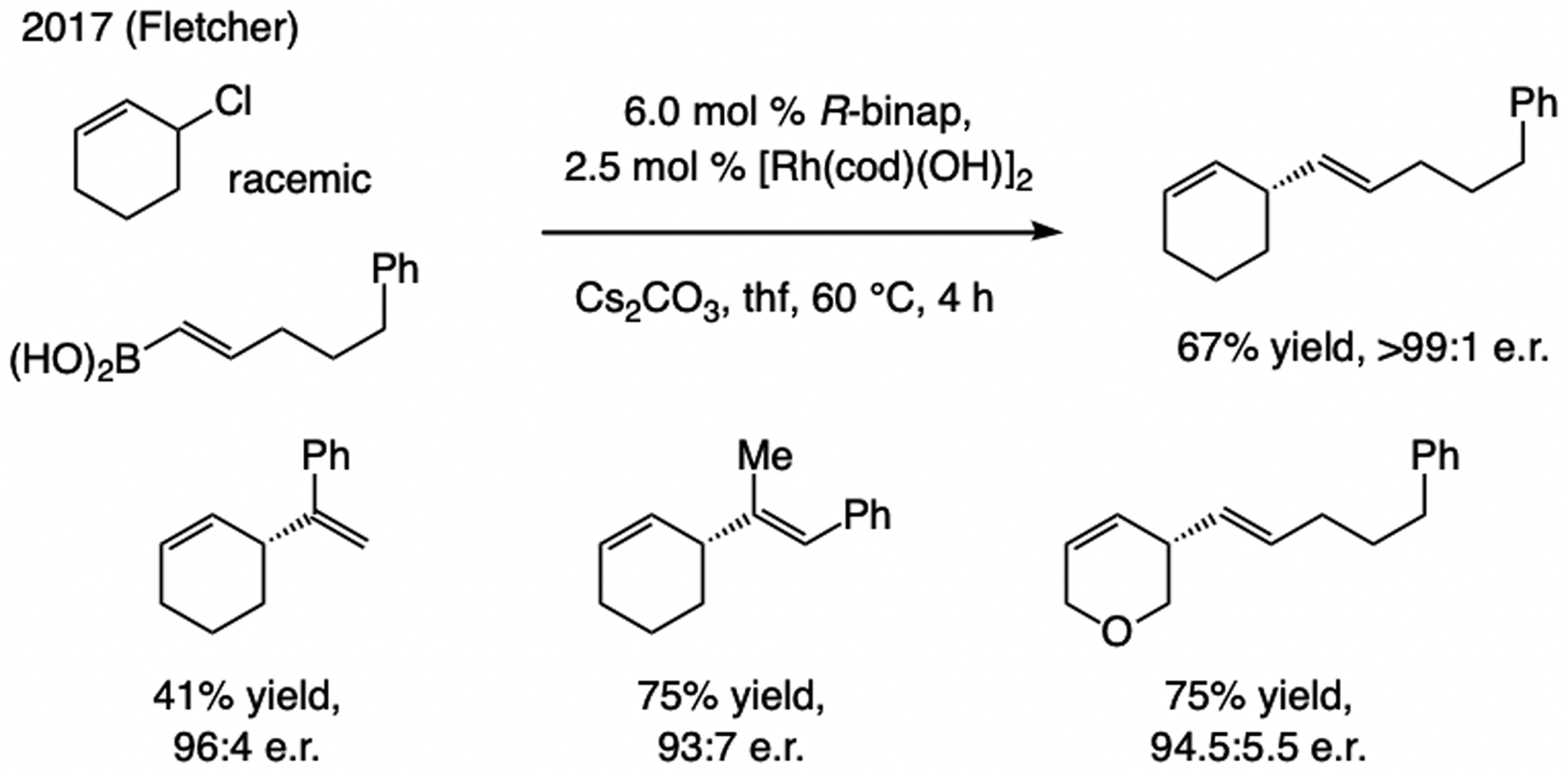
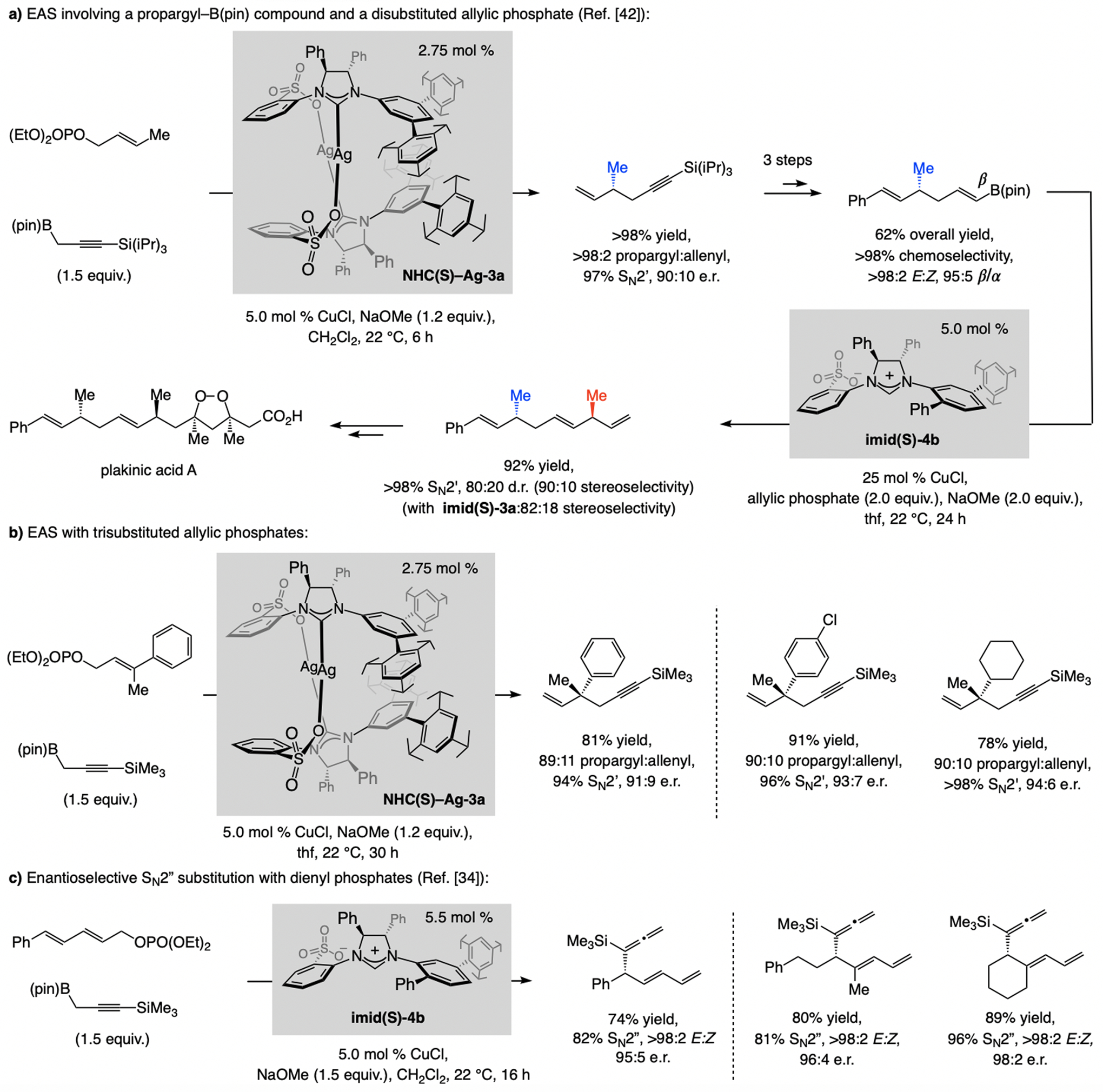
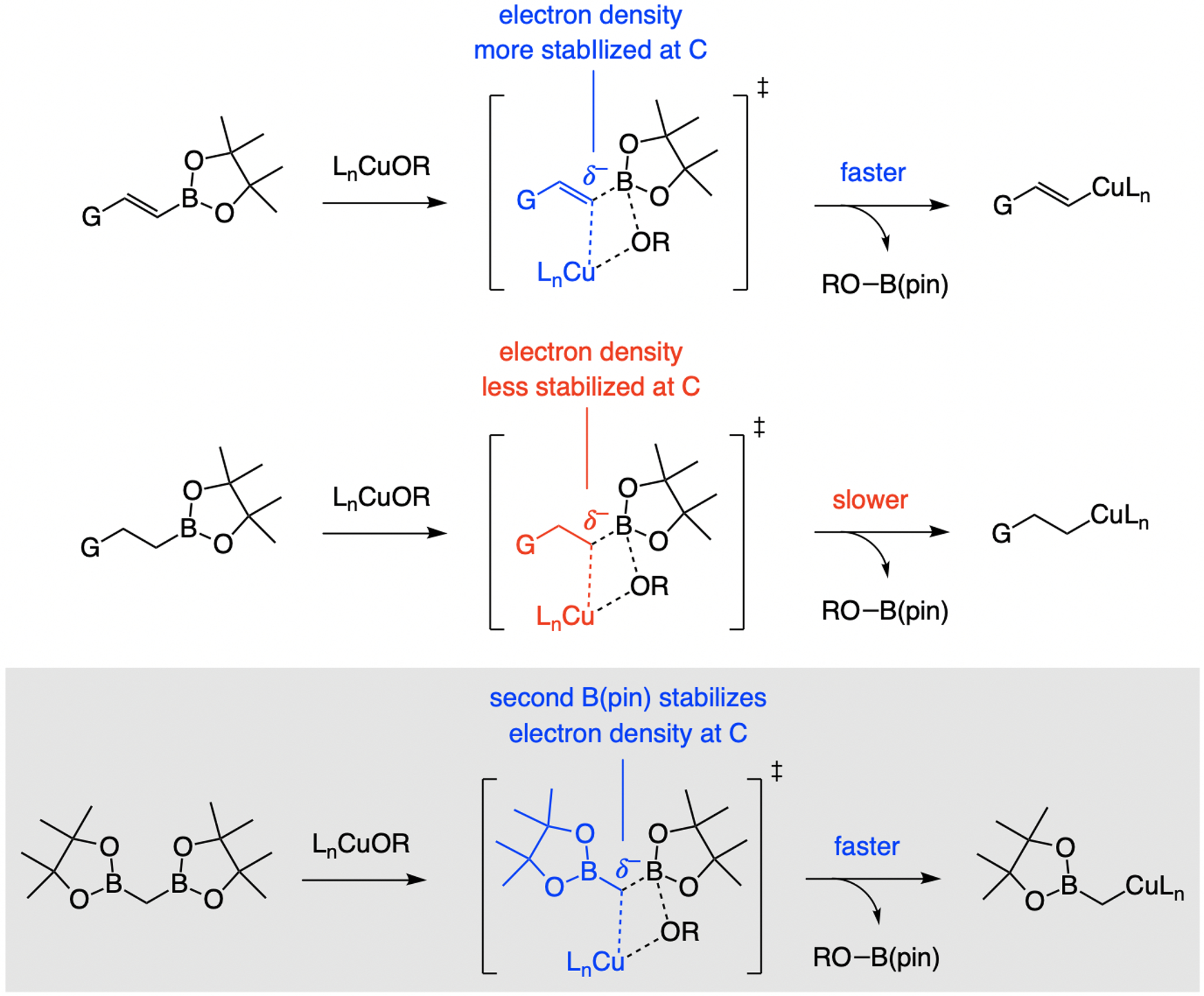
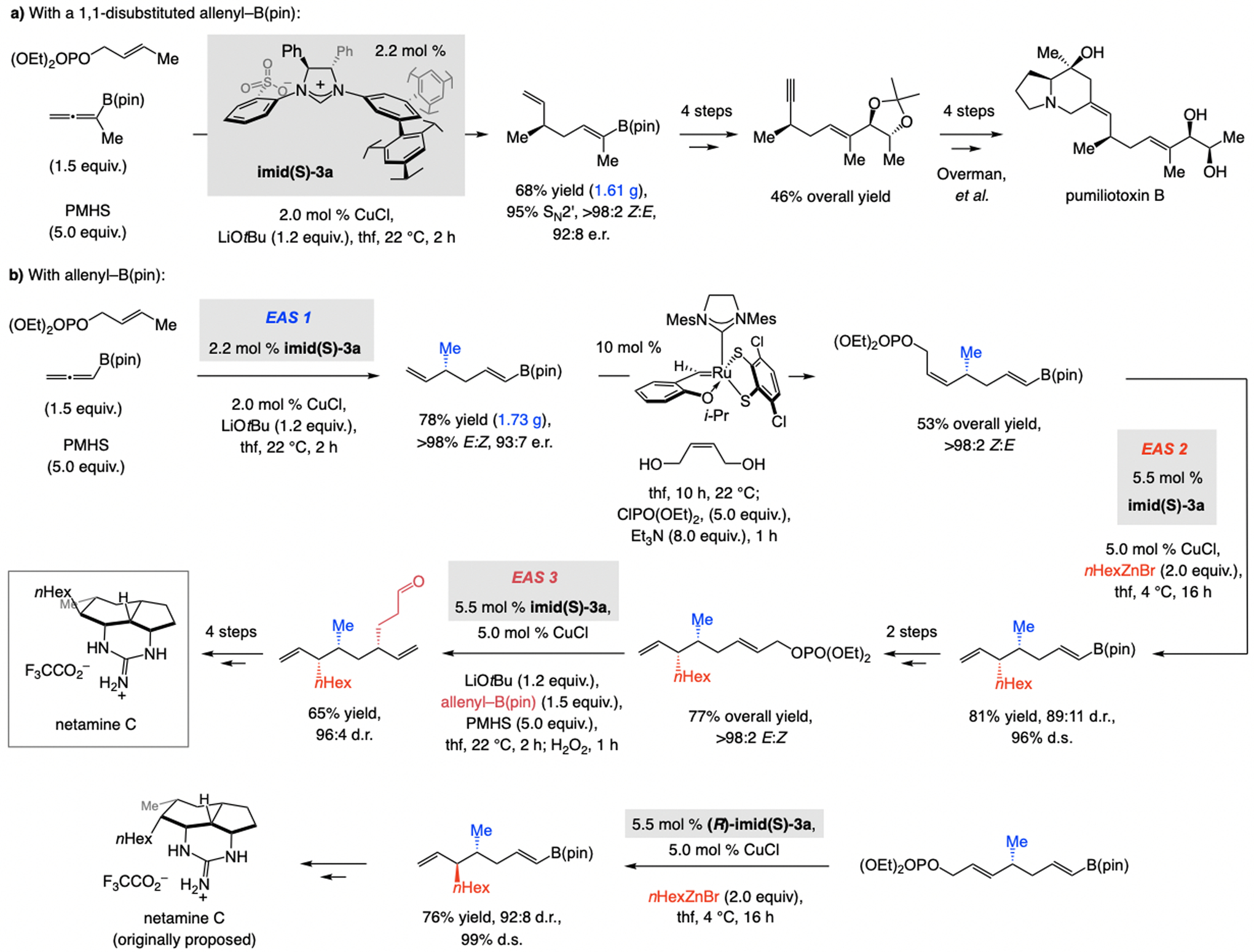
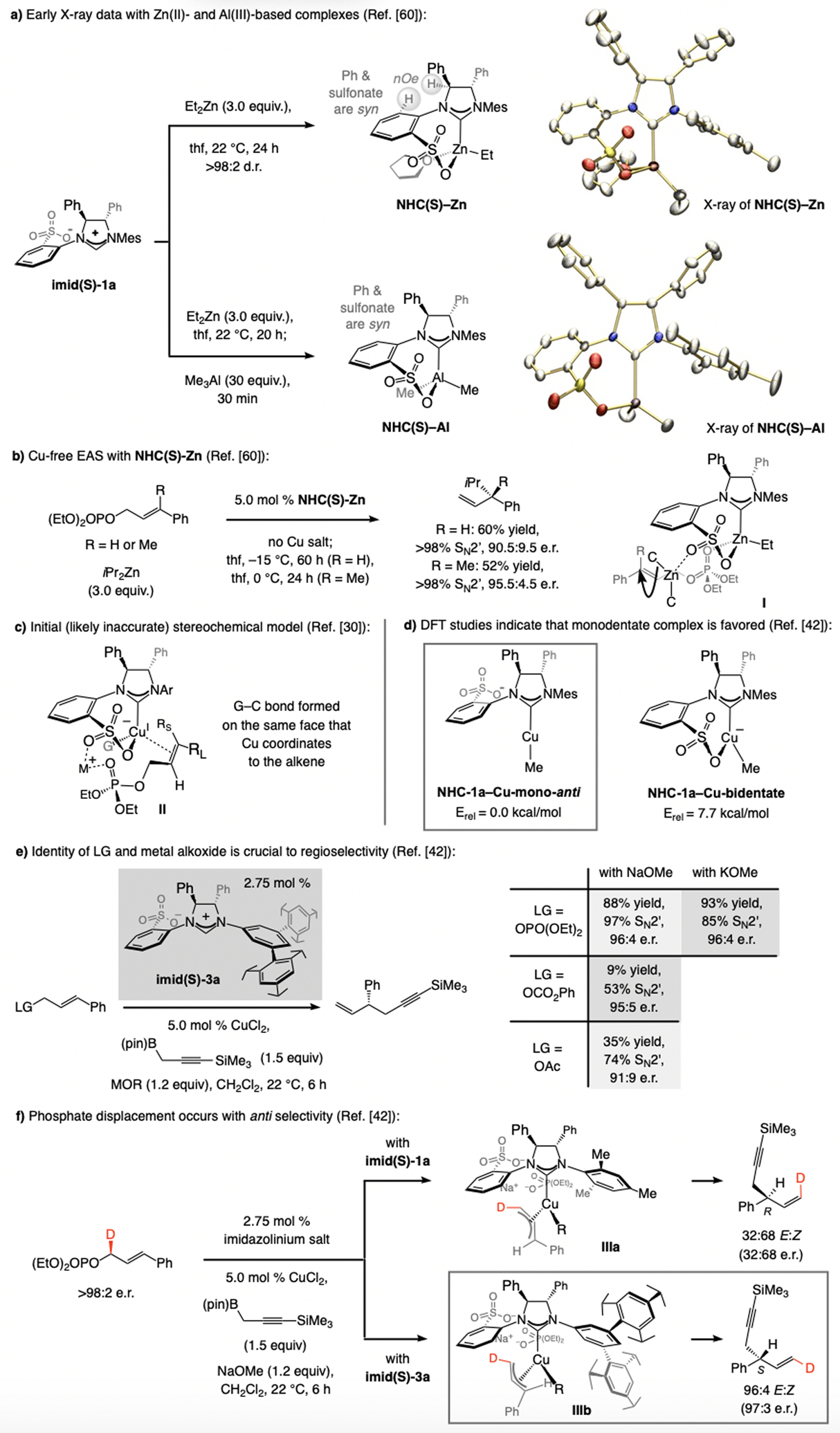
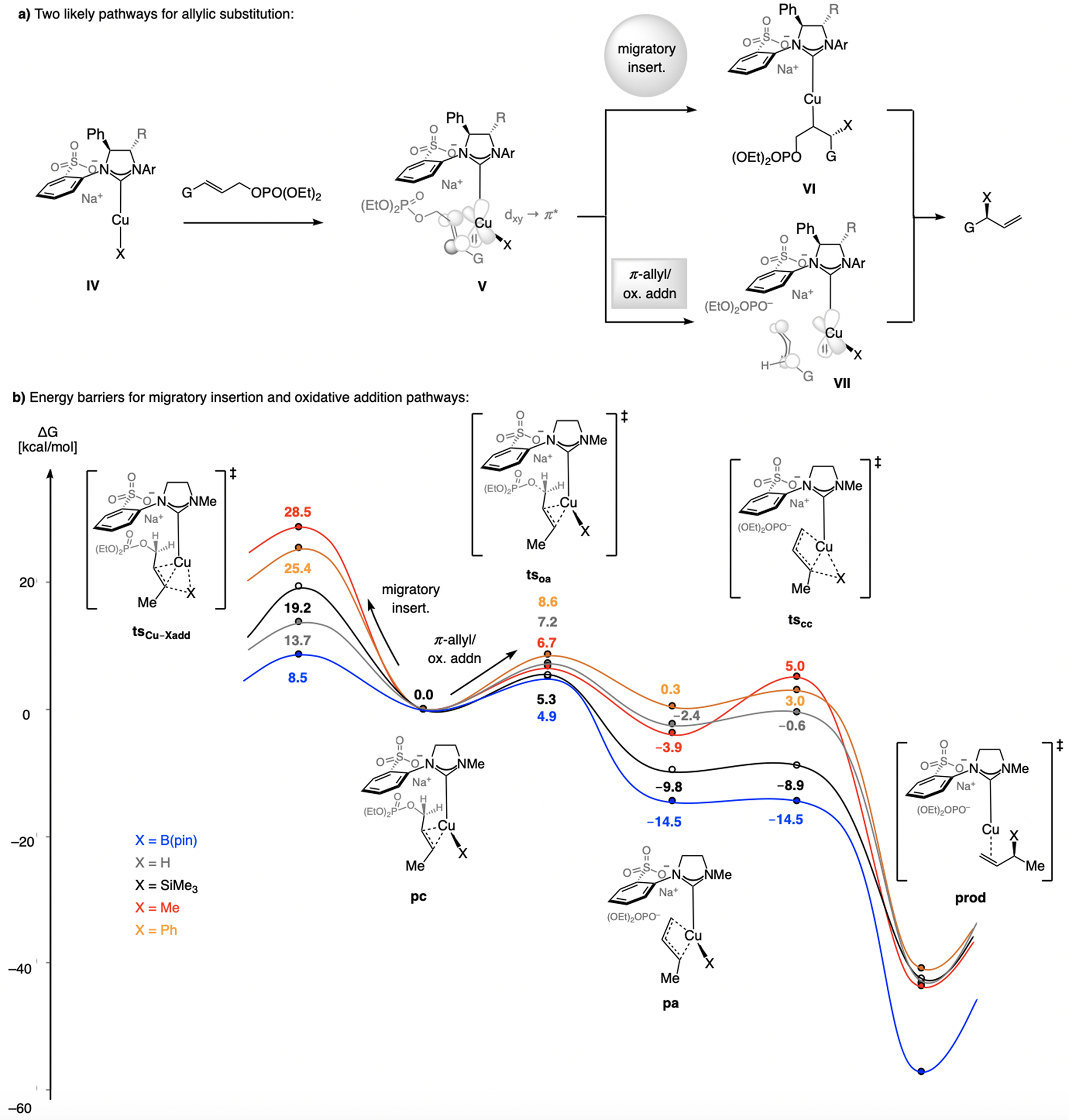
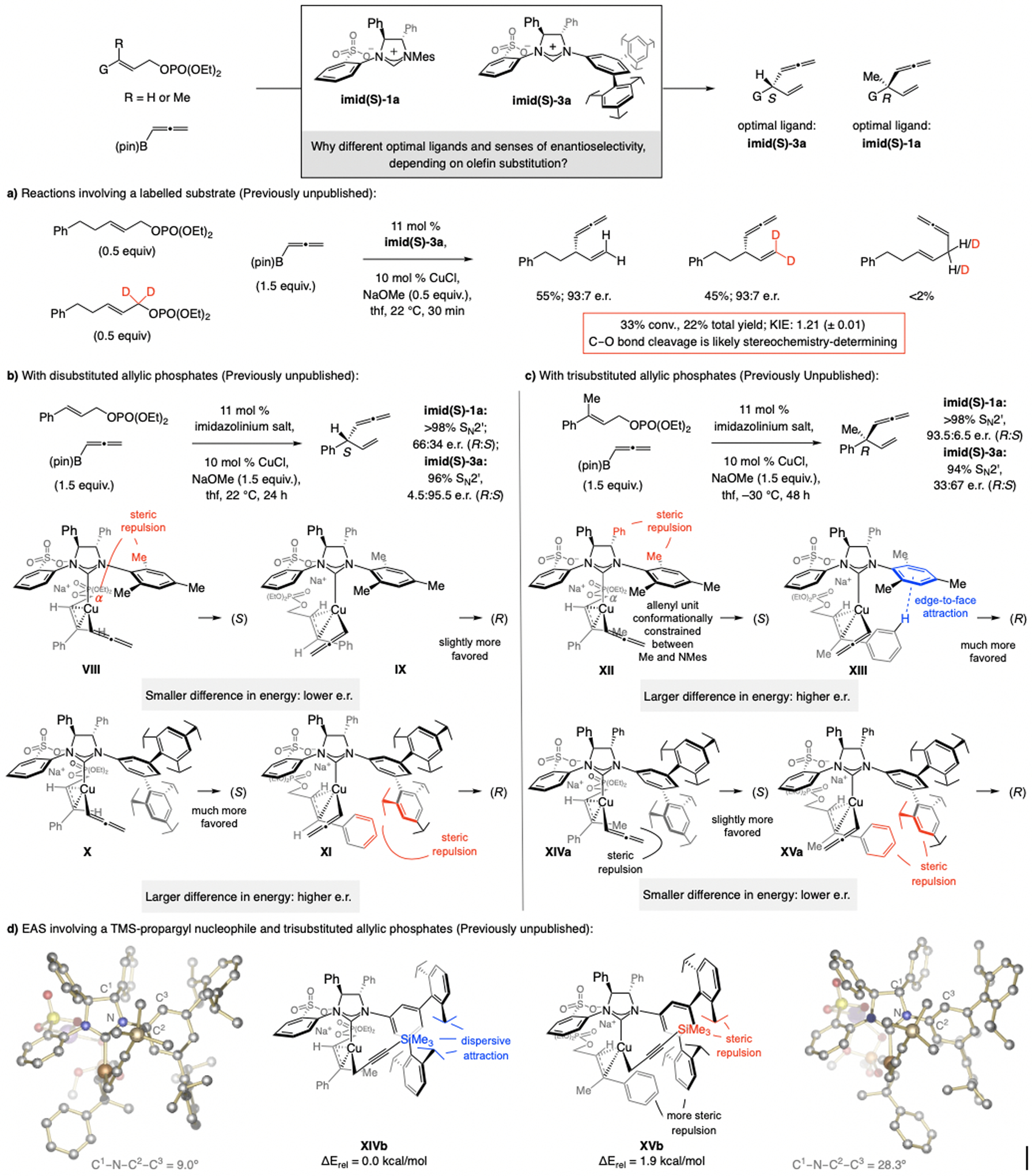
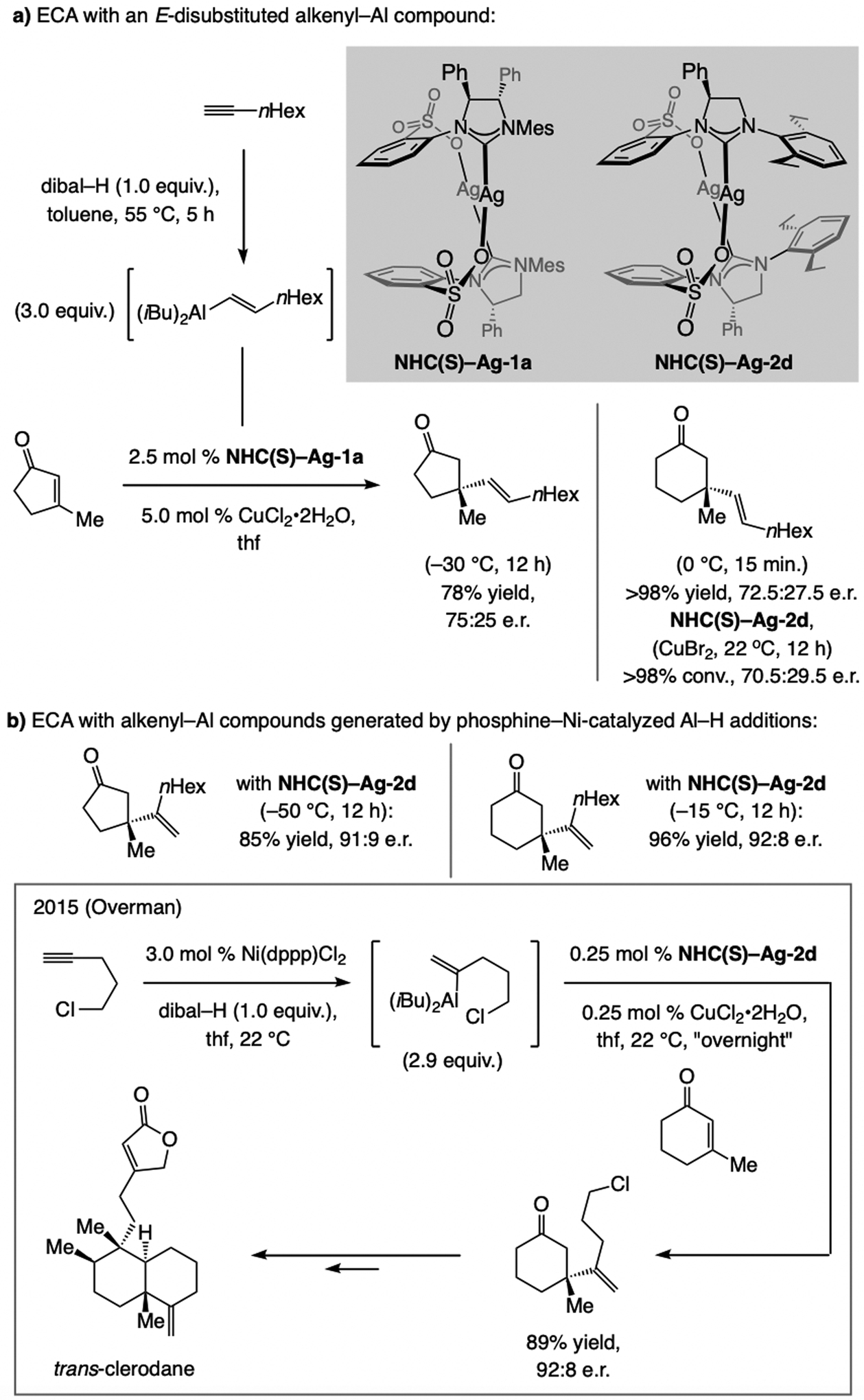
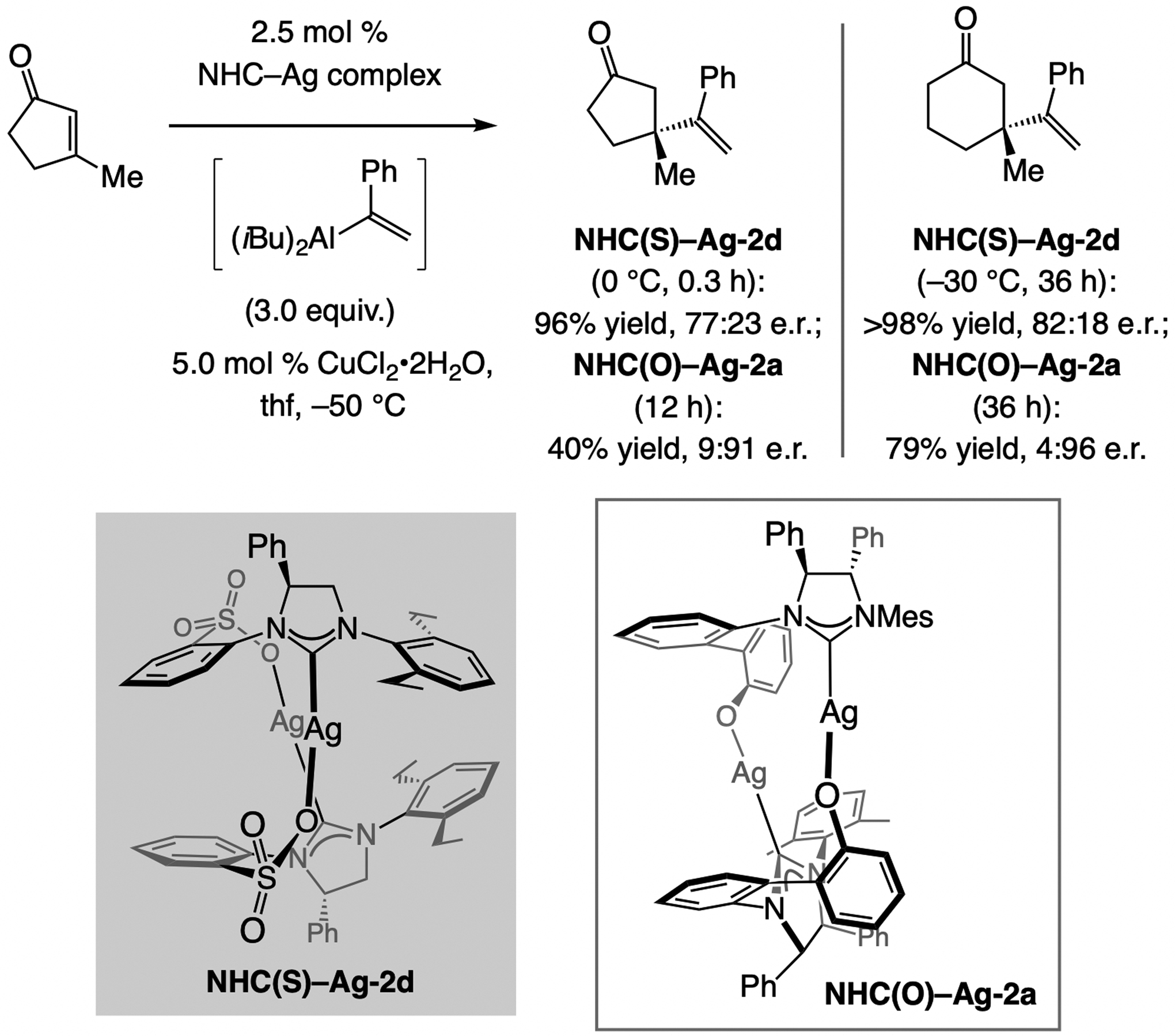
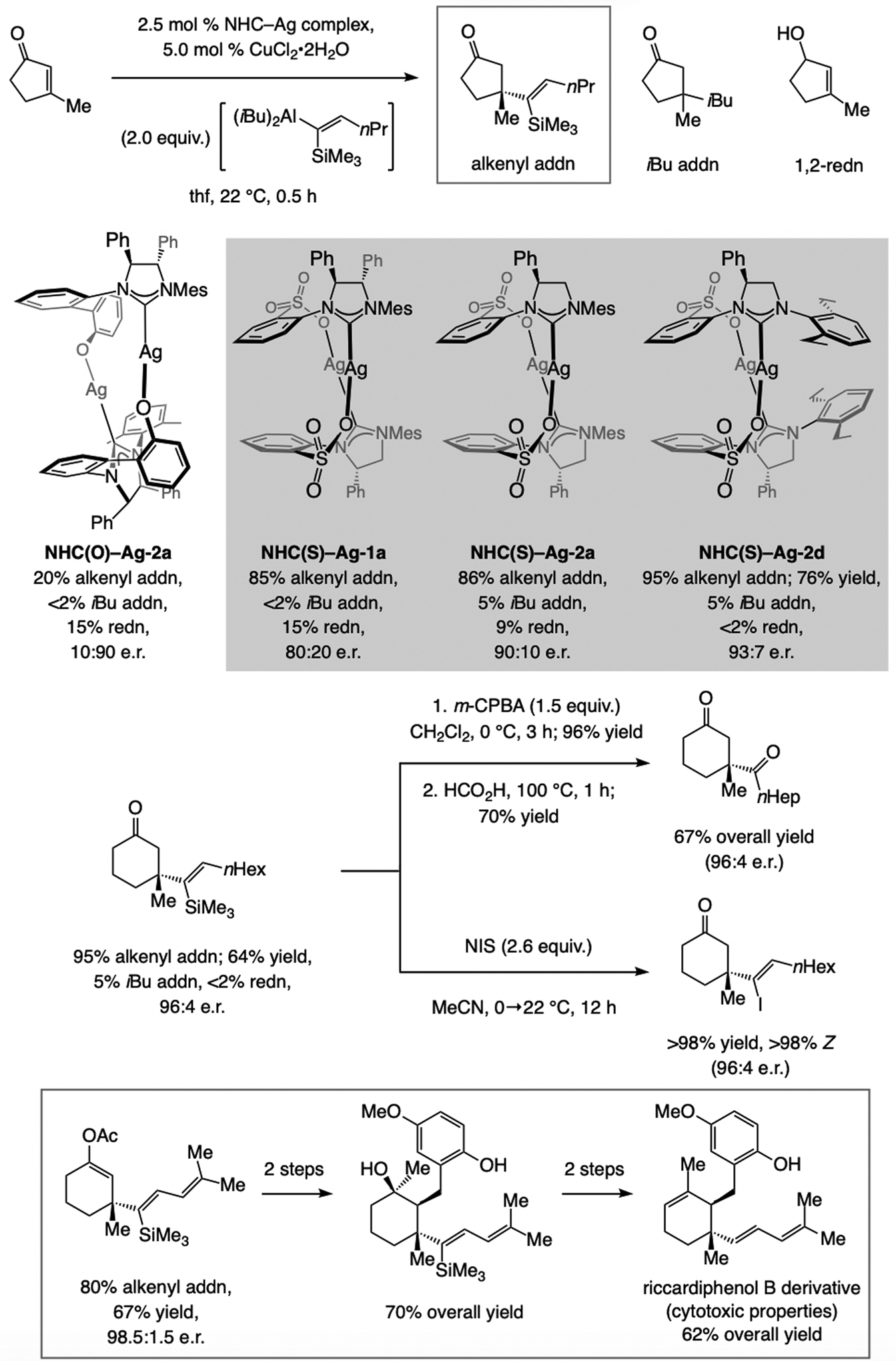
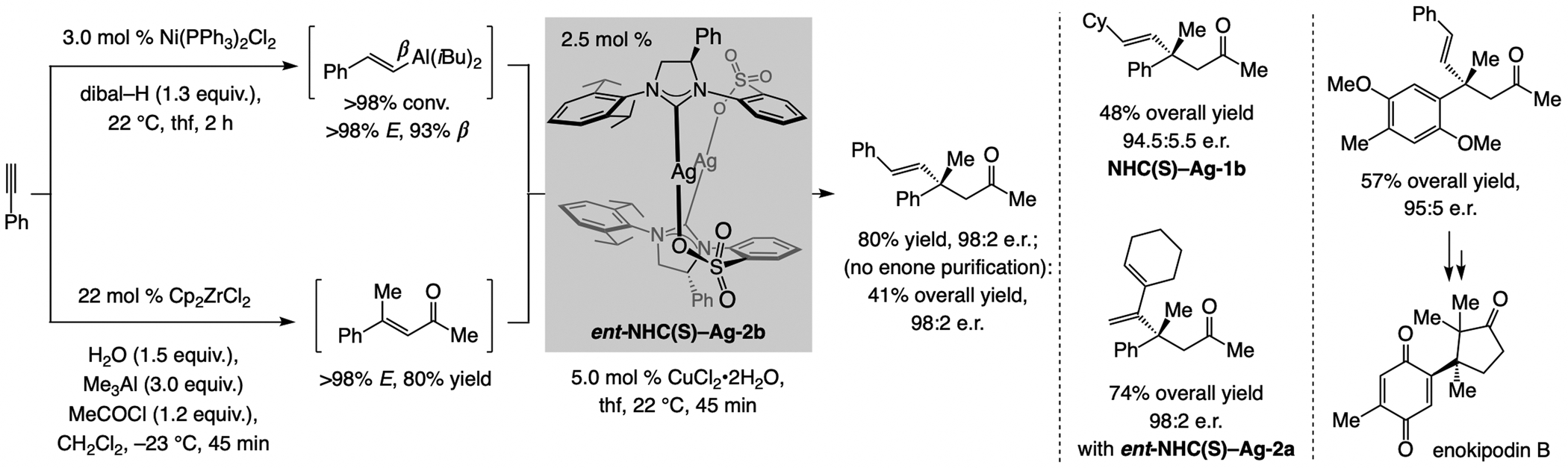
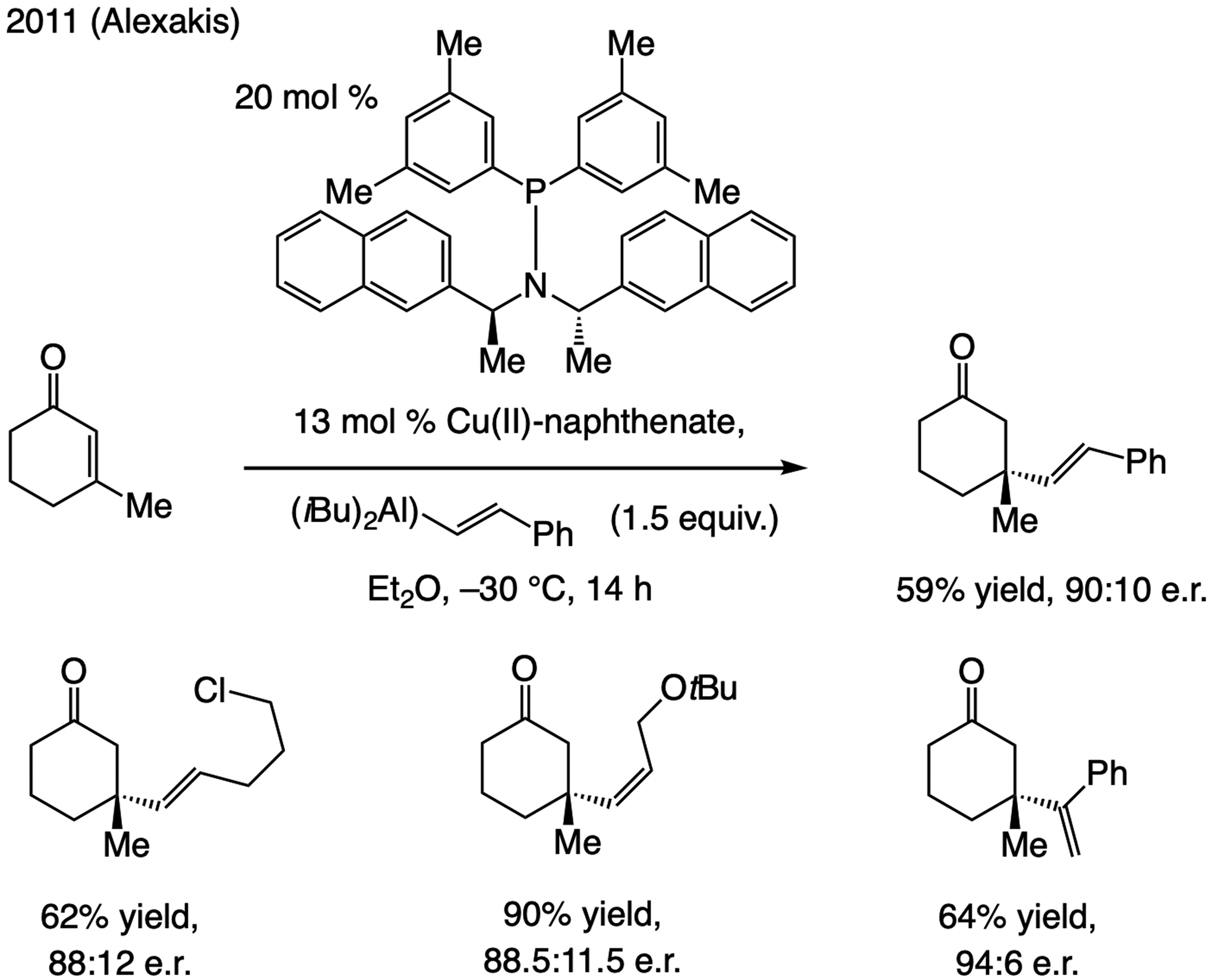
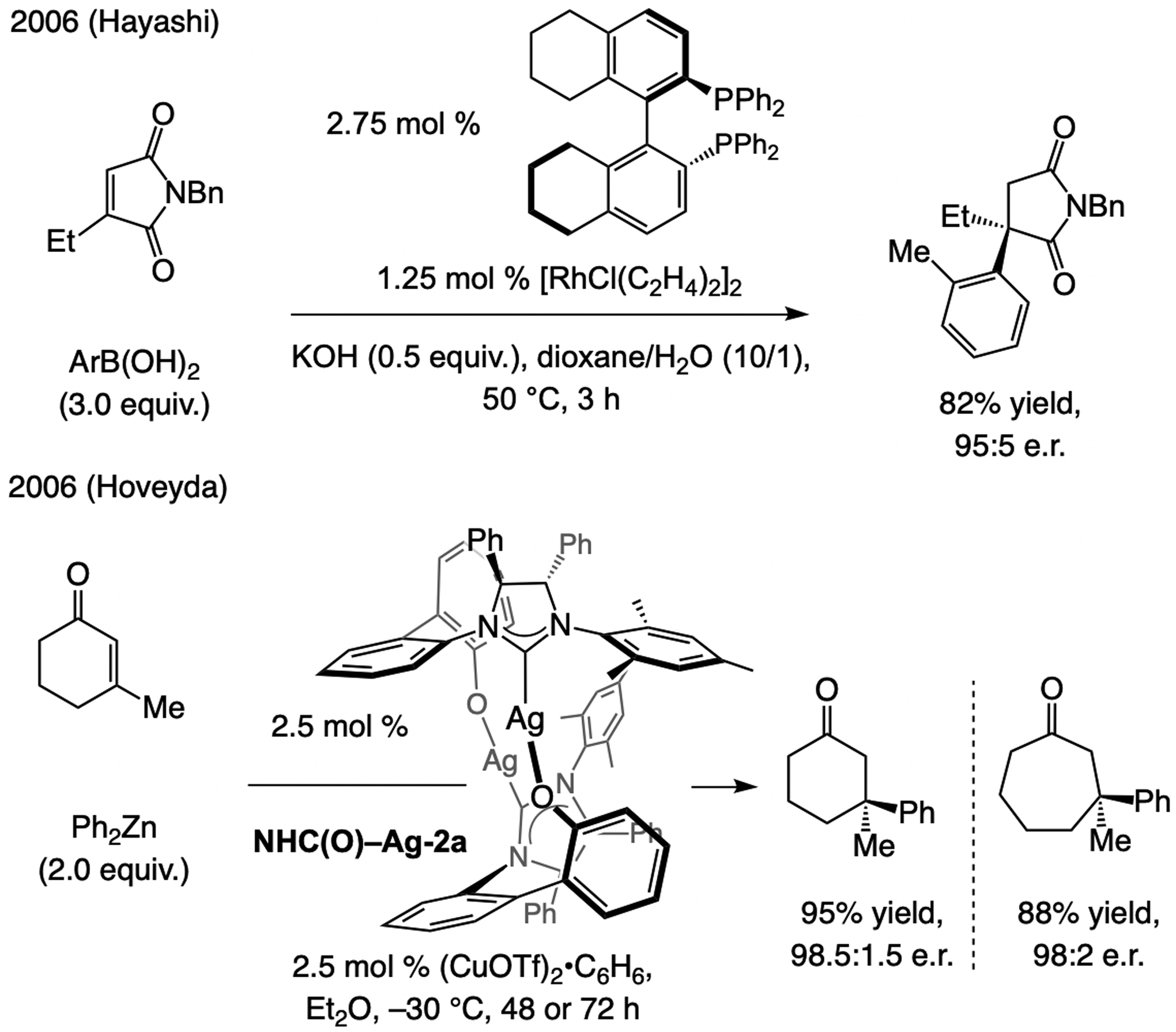
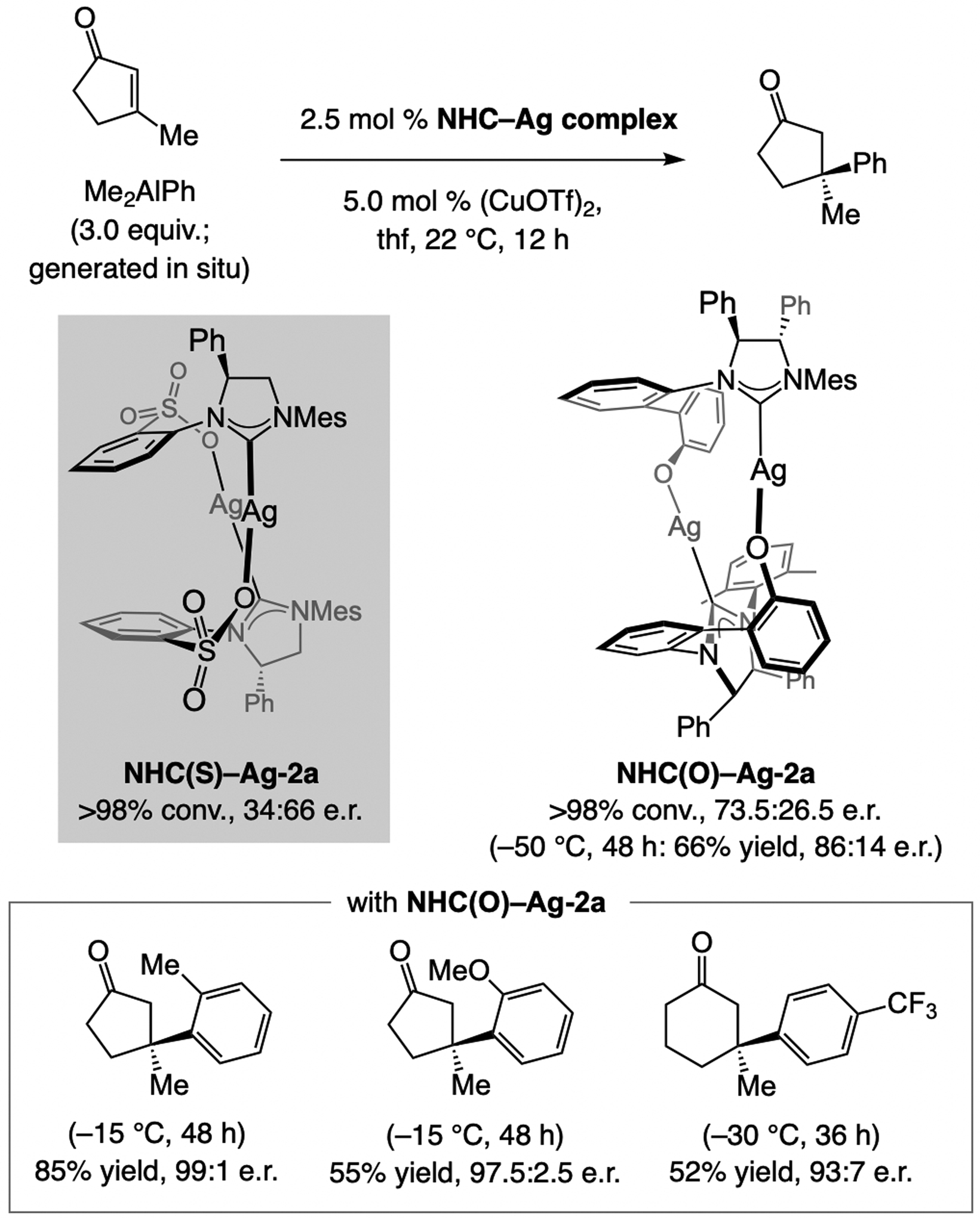
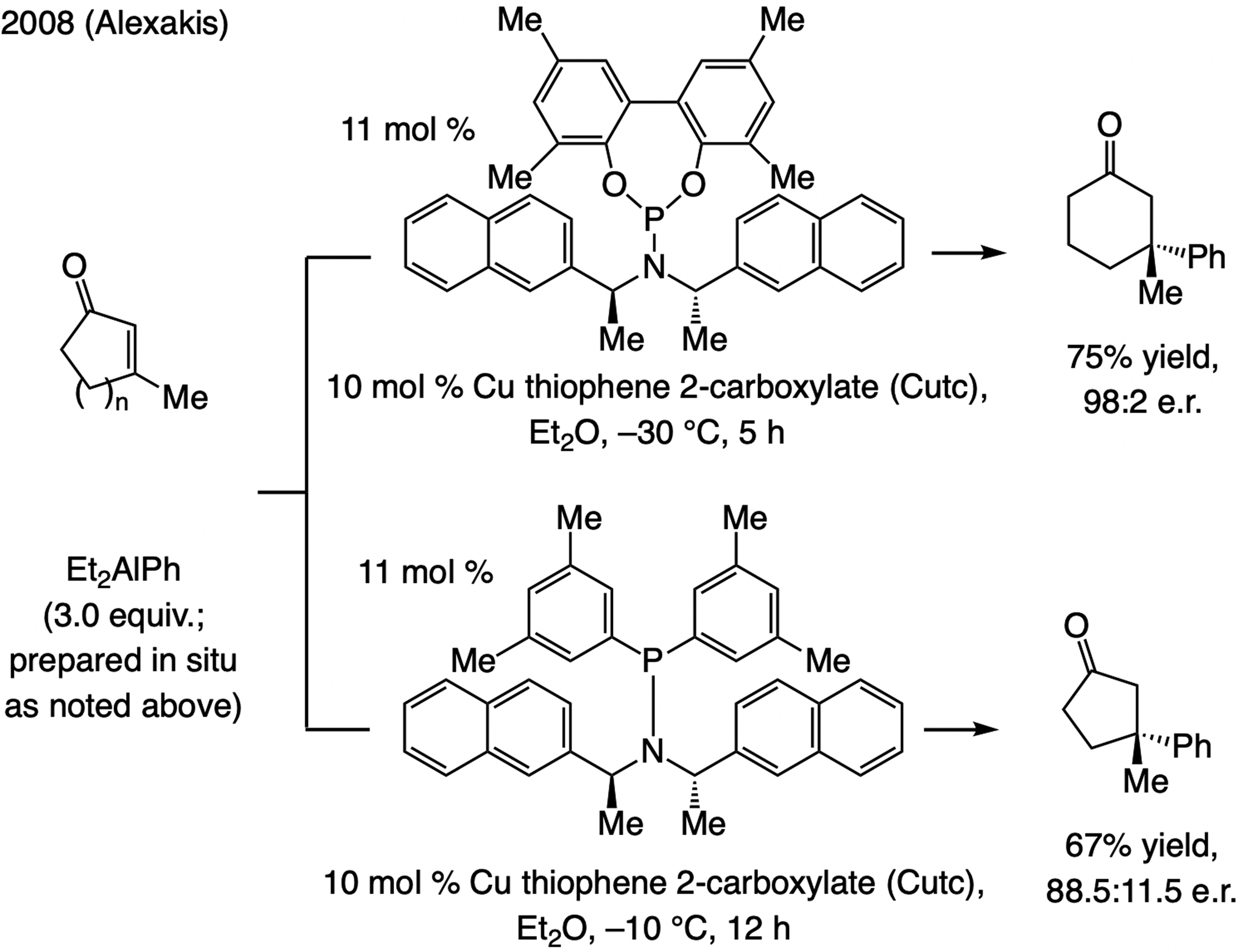

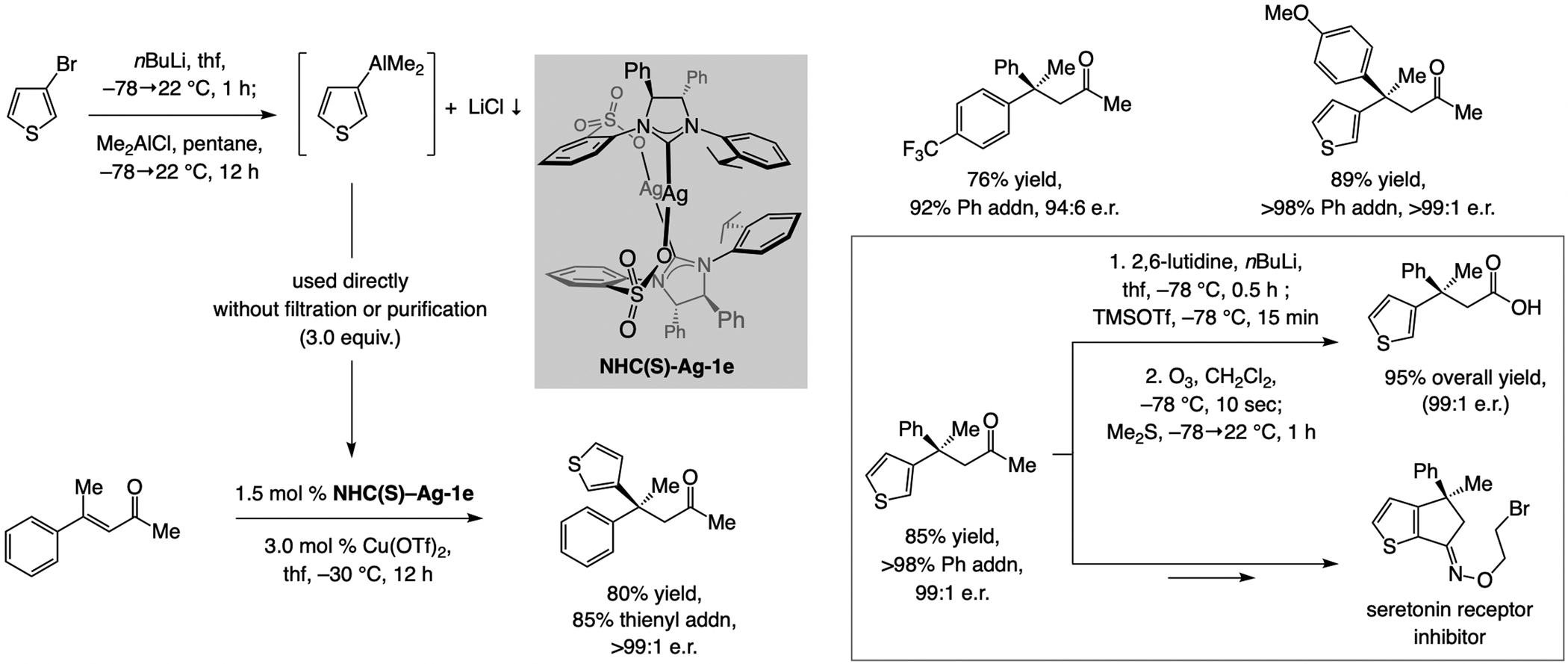
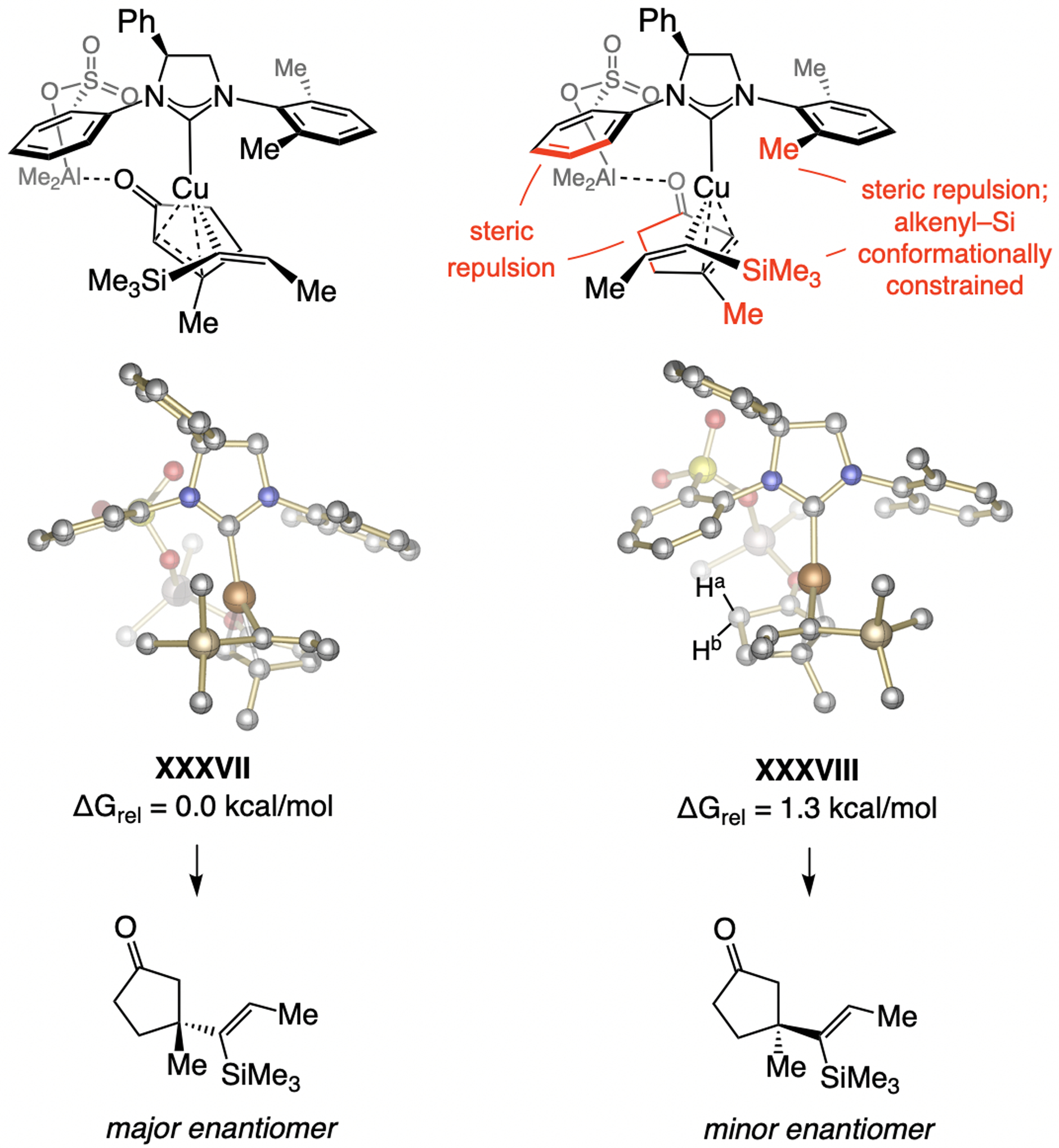
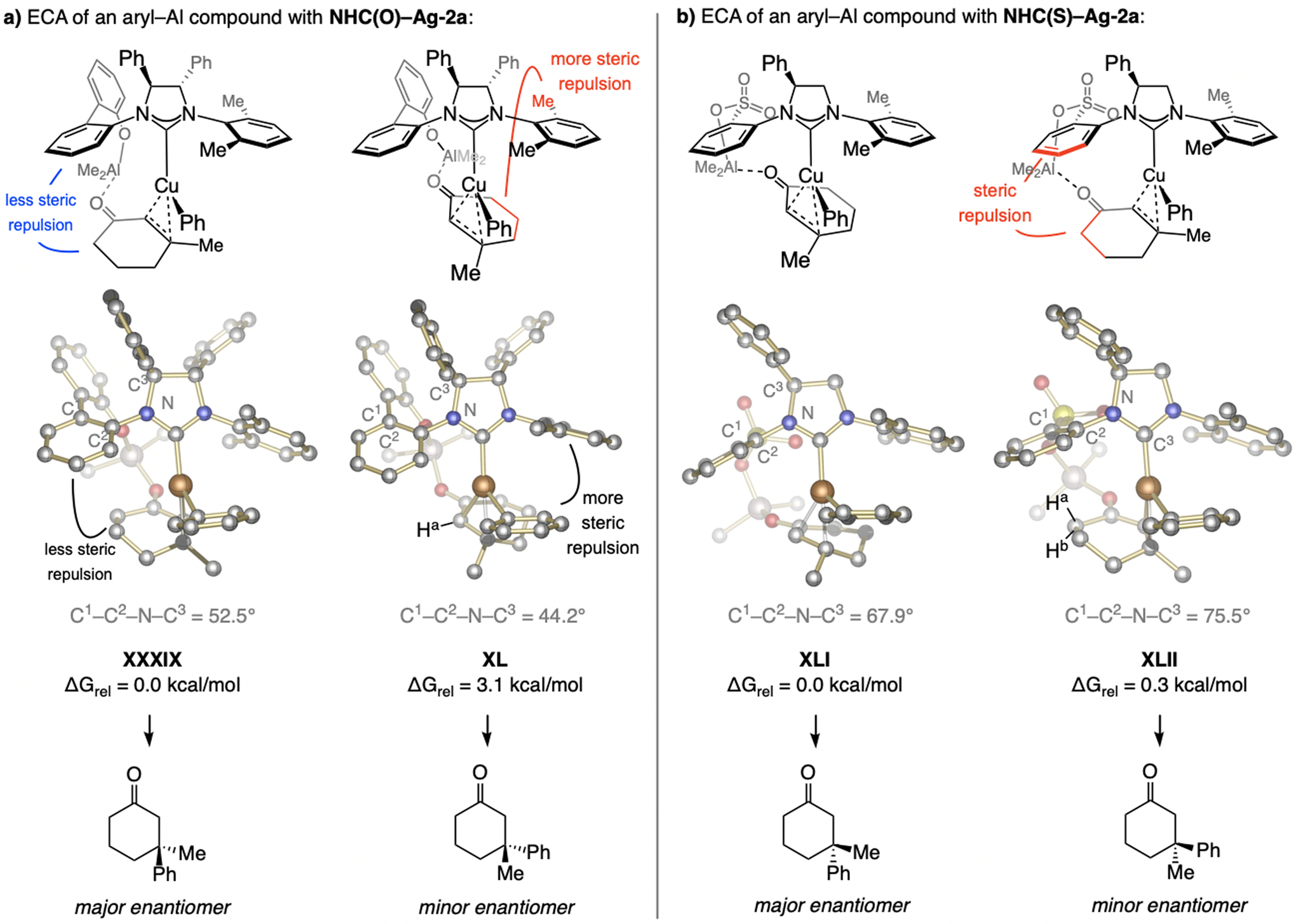
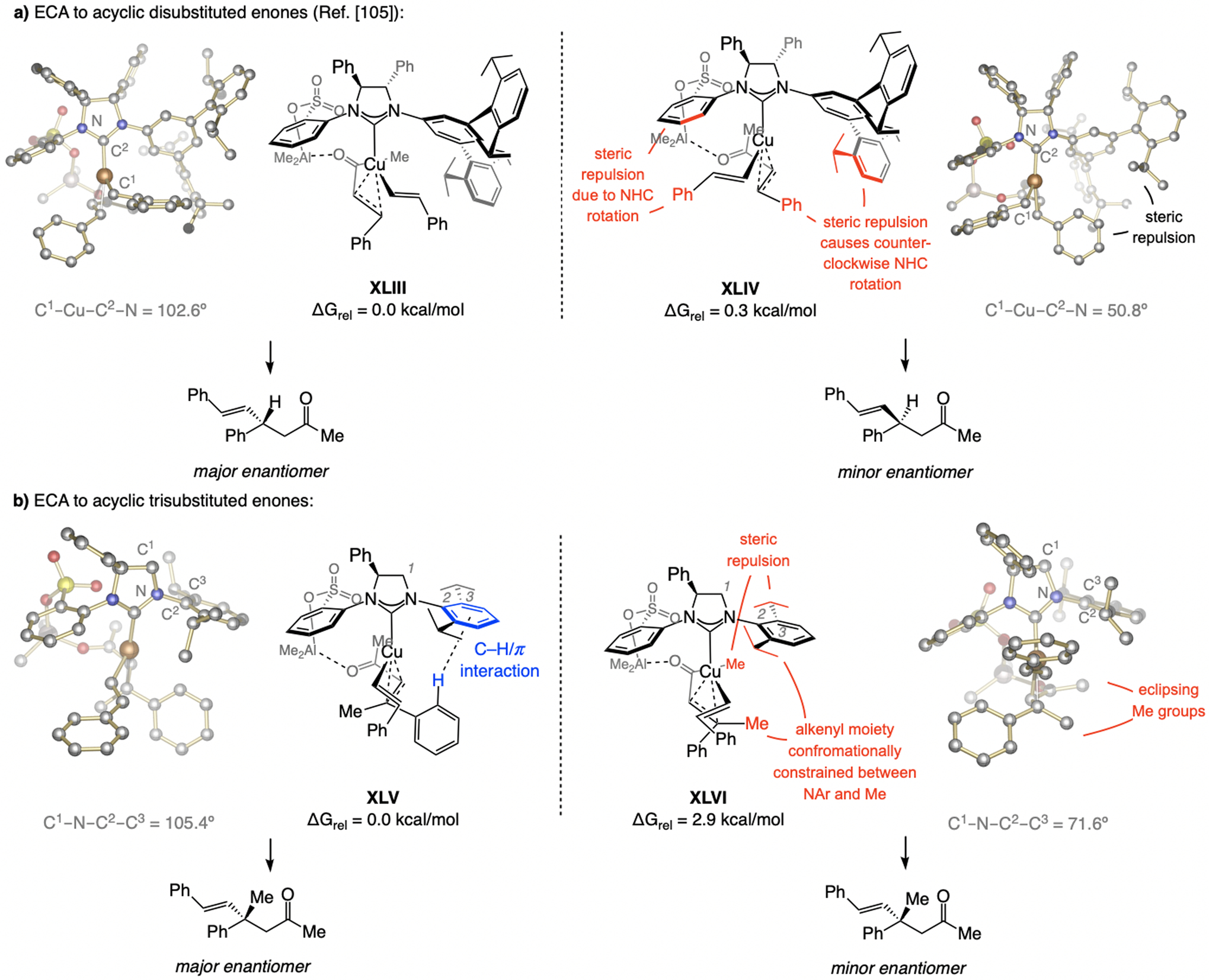
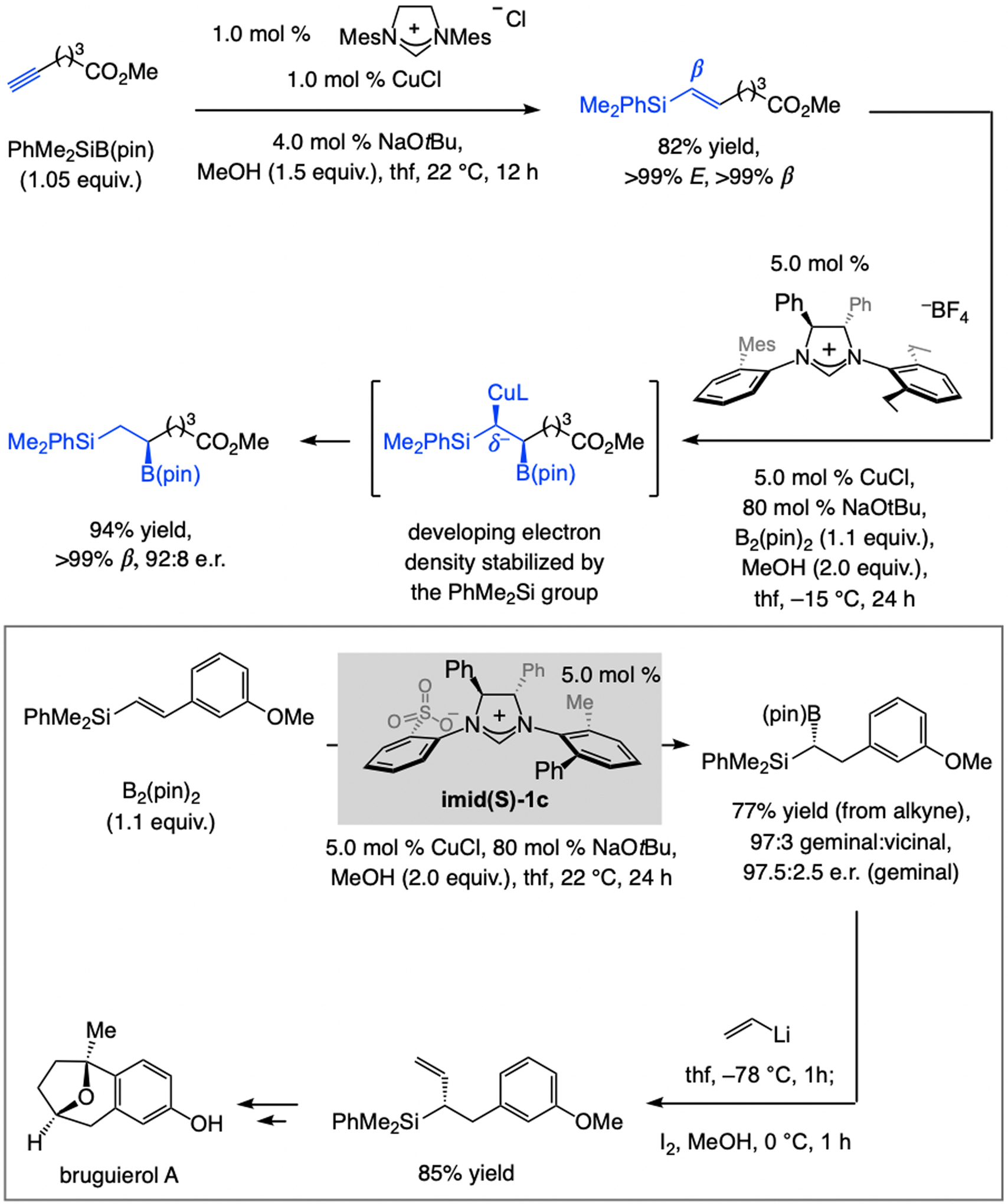
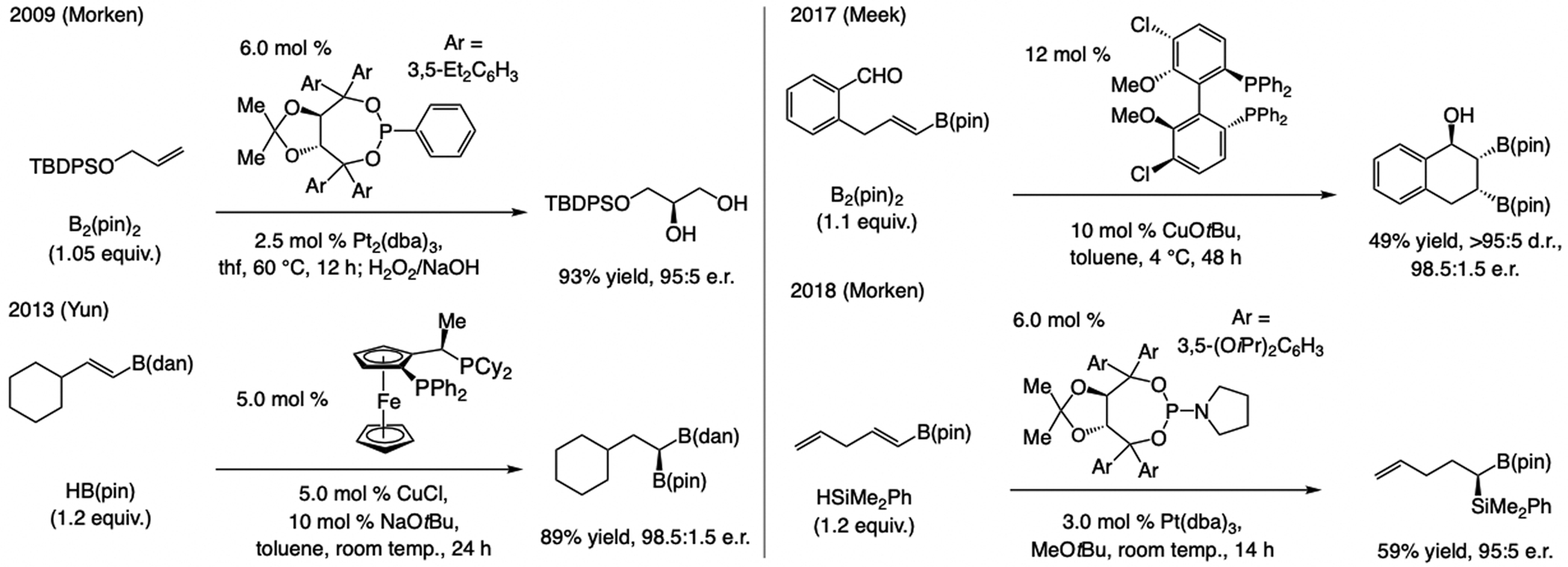
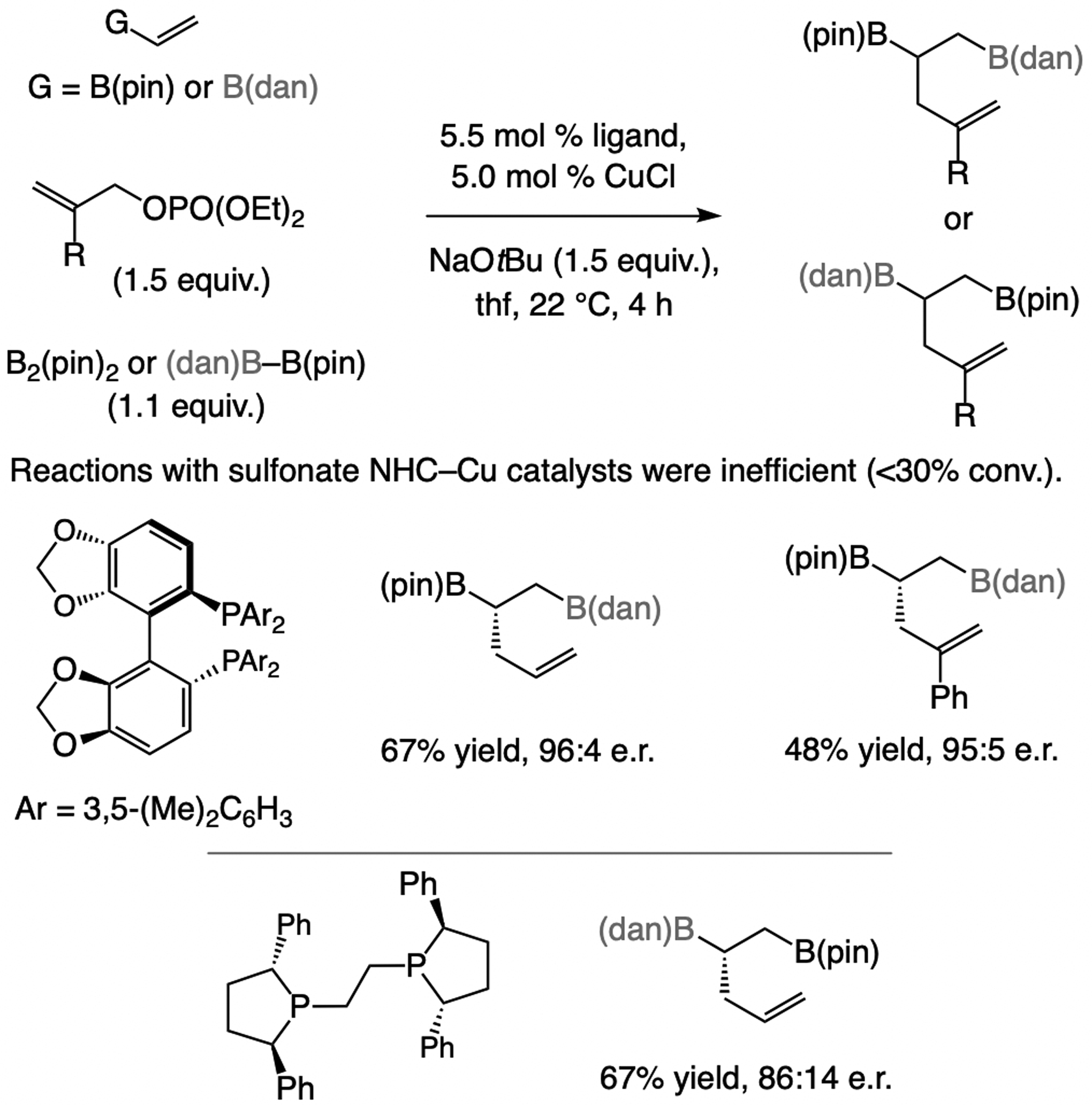
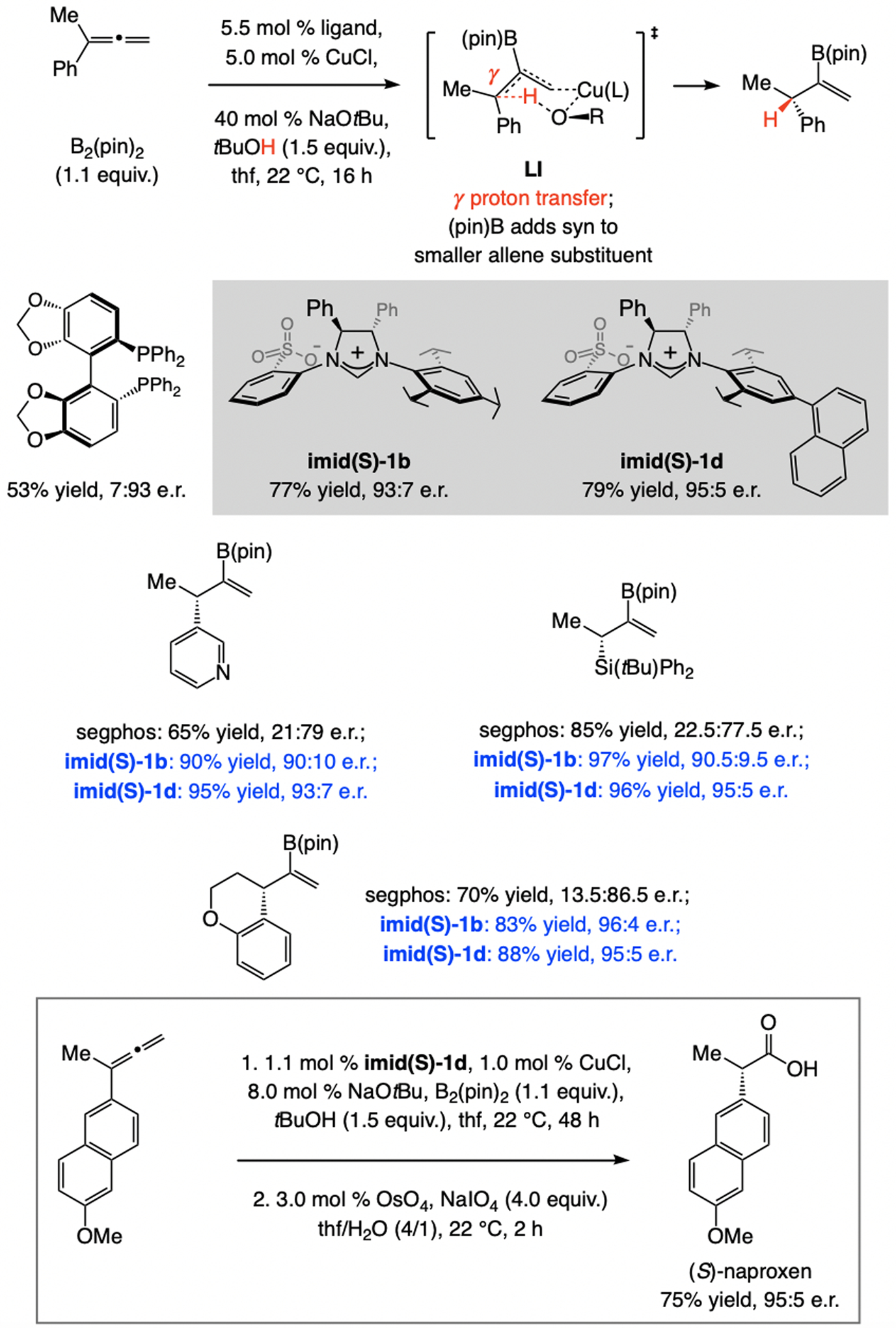
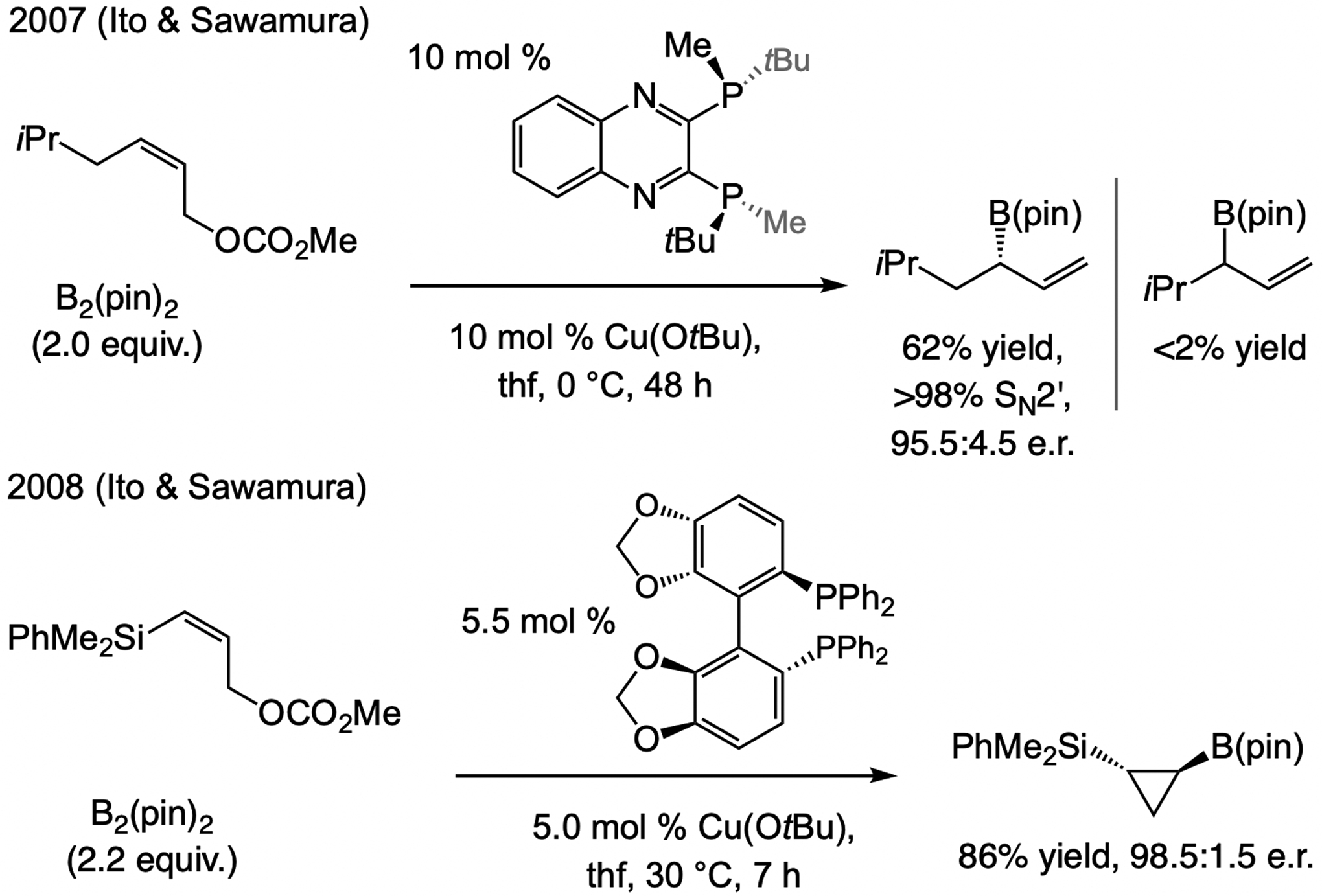
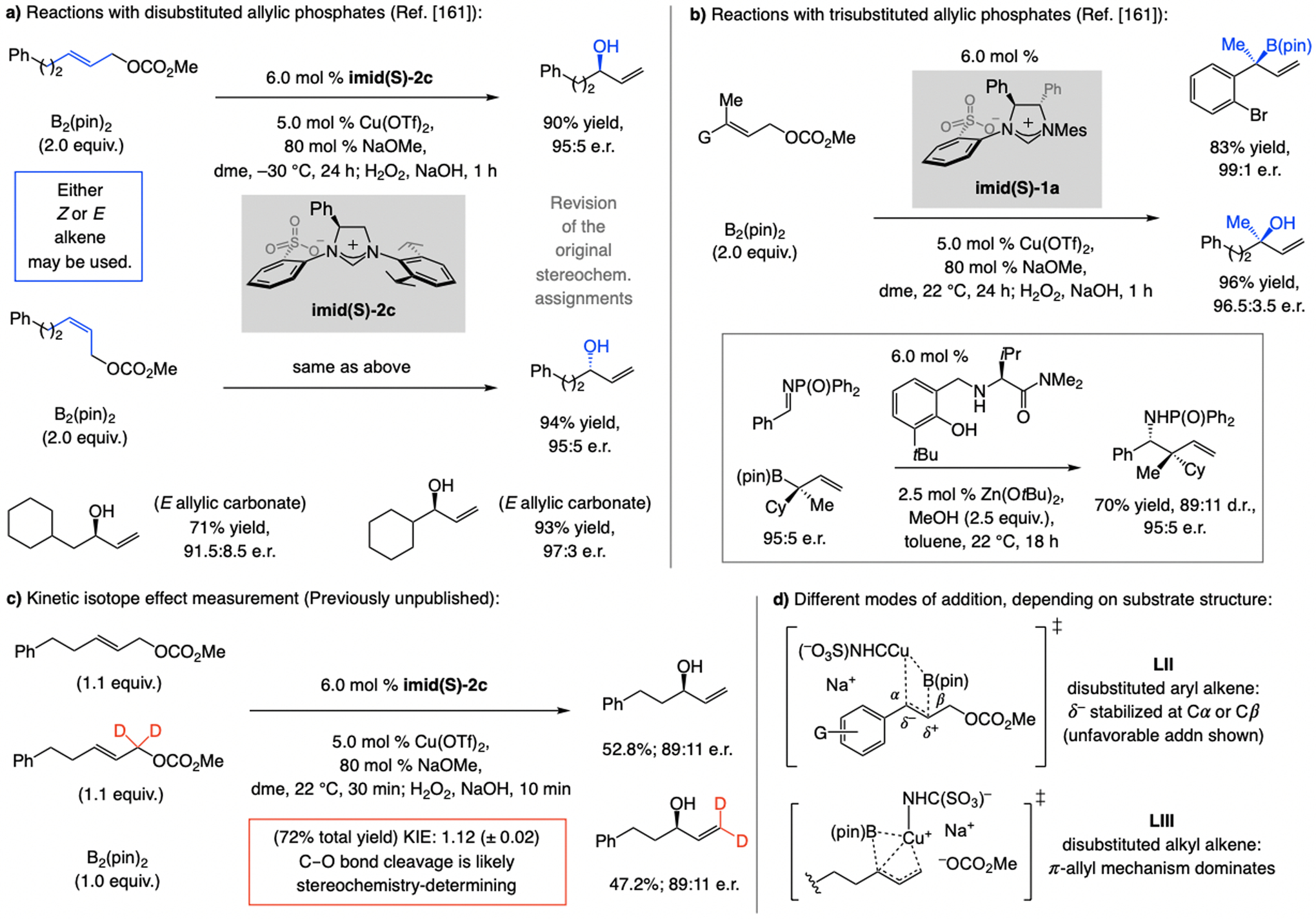
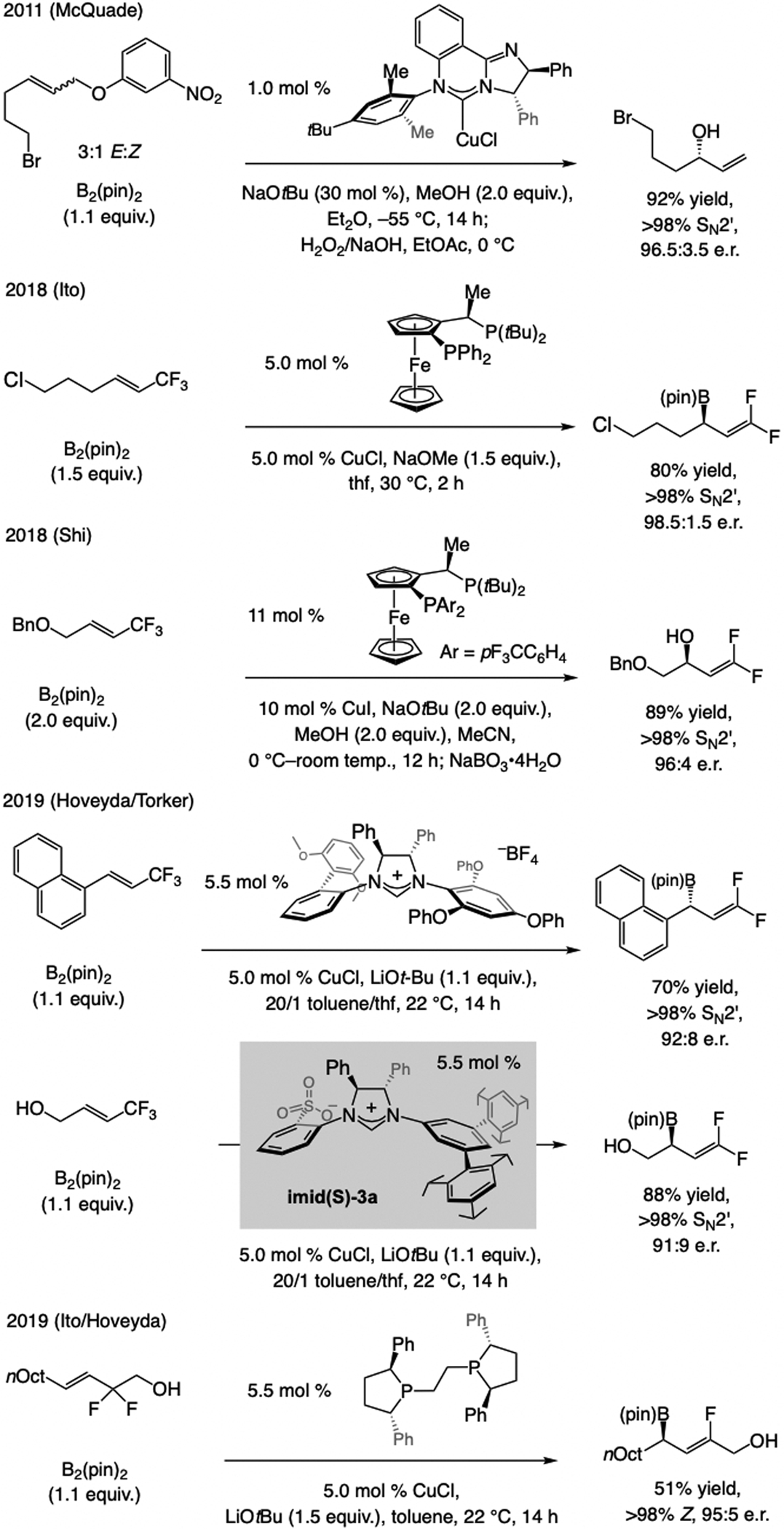
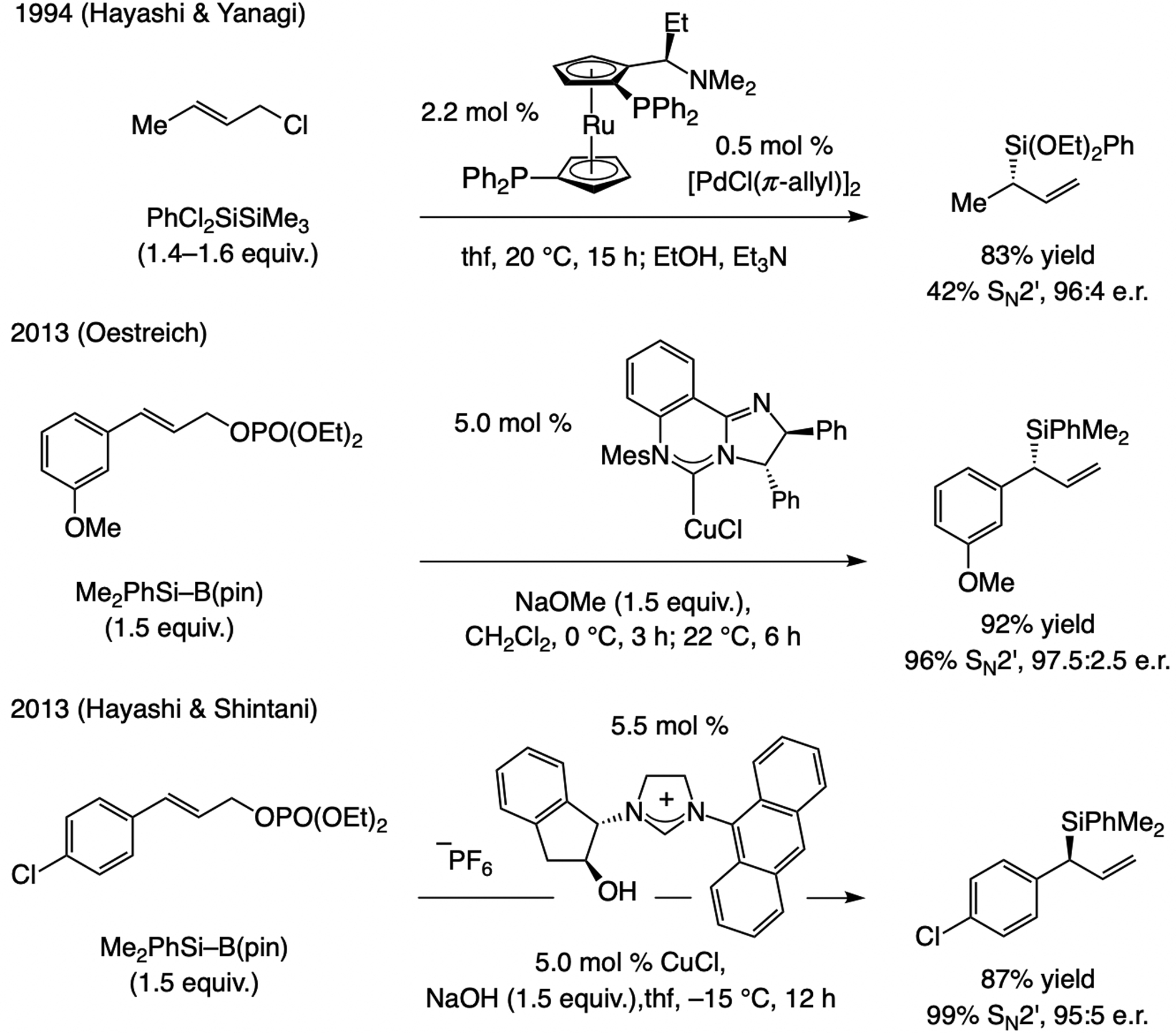
References
-
- van Veldhuizen JJ, Garber SB, Kingsbury JS, Hoveyda AH, J. Am. Chem. Soc 2002, 124, 4954–4955; - PubMed
- van Veldhuizen JJ, Gillingham DG, Garber SB, Kataoka O, Hoveyda AH, J. Am. Chem. Soc 2003, 125, 12502–12508; - PubMed
- Gillingham DG, Kataoka O, Garber SB, Hoveyda AH, J. Am. Chem. Soc 2004, 126, 12288–12290. - PubMed
-
- Larsen AO, Leu W, Oberhuber C. Nieto, Campbell JE, Hoveyda AH, J. Am. Chem. Soc 2004, 126, 11130–11131; - PubMed
- Van Veldhuizen JJ, Campbell JE, Guidici RE, Hoveyda AH, J. Am. Chem. Soc 2005, 127, 6877–6882; - PubMed
- Lee Y, Hoveyda AH, J. Am. Chem. Soc 2006, 128, 15604–15605; - PubMed
- Kacprzynski MA, May TL, Kazane SA, Hoveyda AH, Angew. Chem. Int. Ed 2007, 46, 4554–4558. - PubMed
-
- Lee K.-s., Brown MK, Hird AW, Hoveyda AH, J. Am. Chem. Soc 2006, 128, 7182–7184. - PubMed
-
- Brown KM, May TL, Baxter CA, Hoveyda AH, Angew. Chem. Int. Ed 2007, 46, 1097–1100. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
