

Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes
- PMID: 32366025
- PMCID: PMC7290700
- DOI: 10.3390/v12050498
Codon Usage and Phenotypic Divergences of SARS-CoV-2 Genes
Abstract
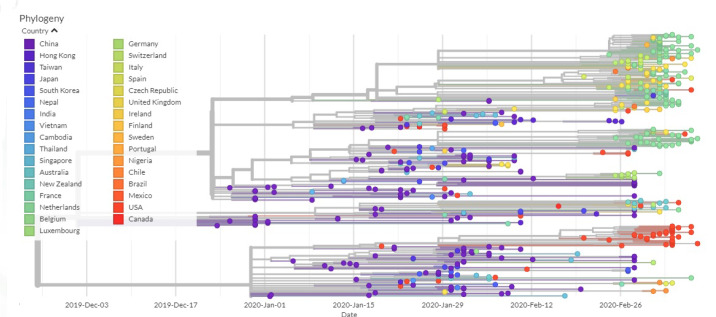
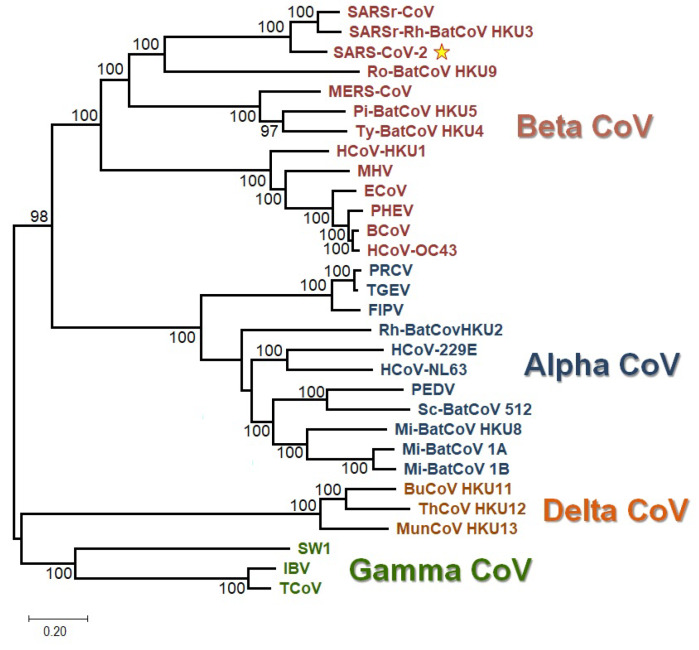
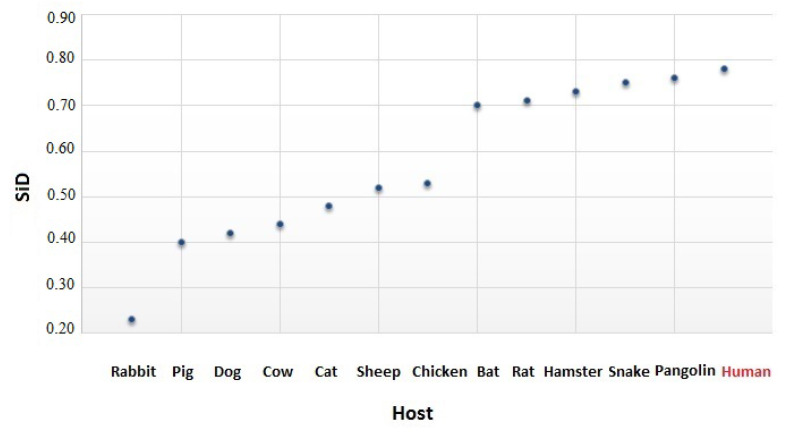
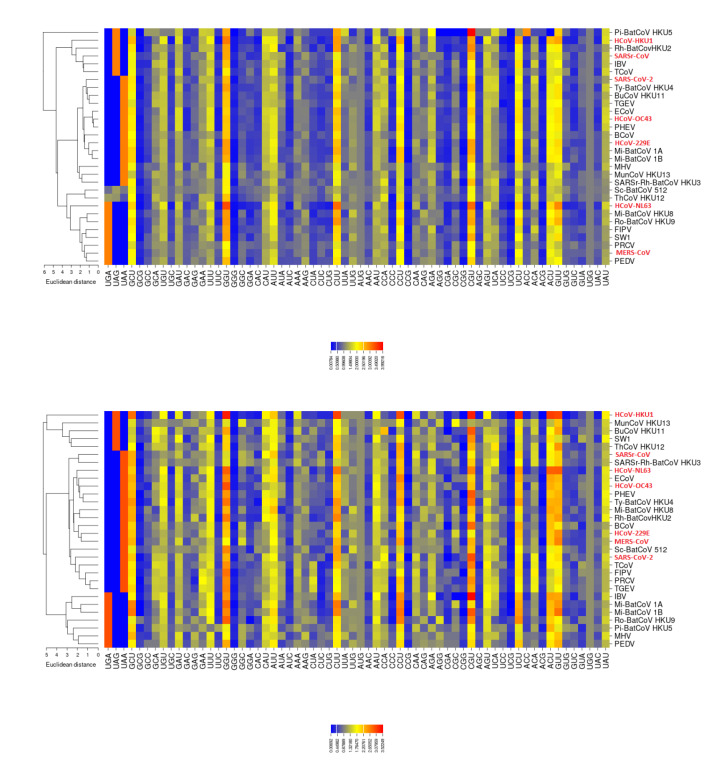
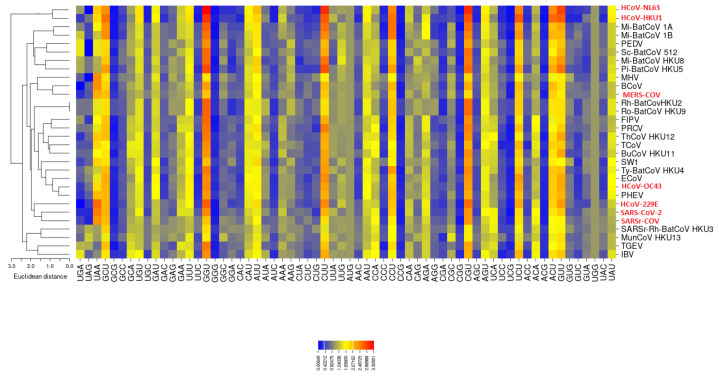
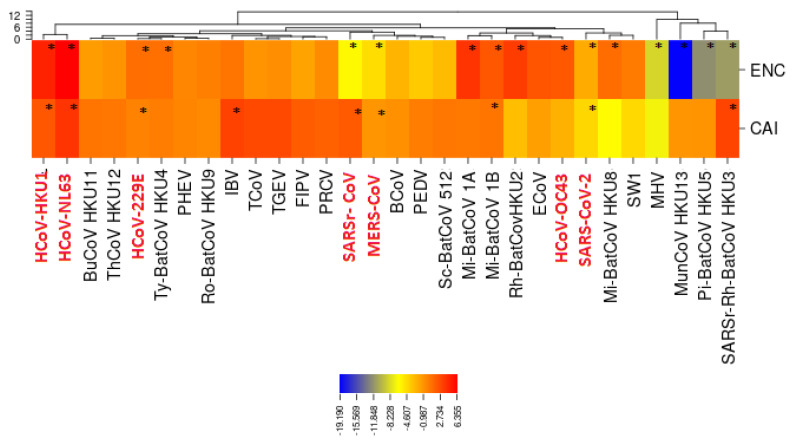
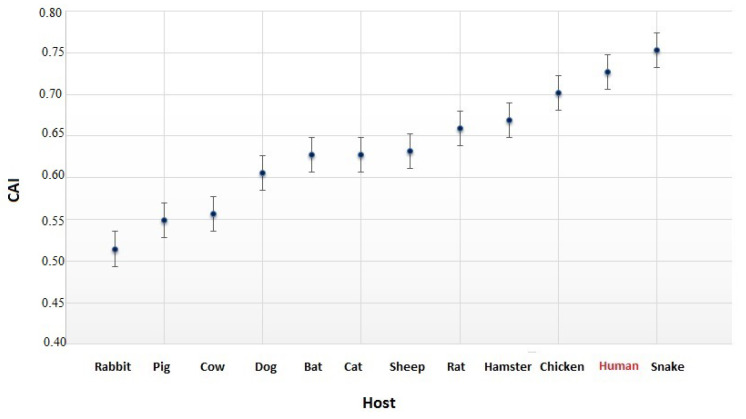
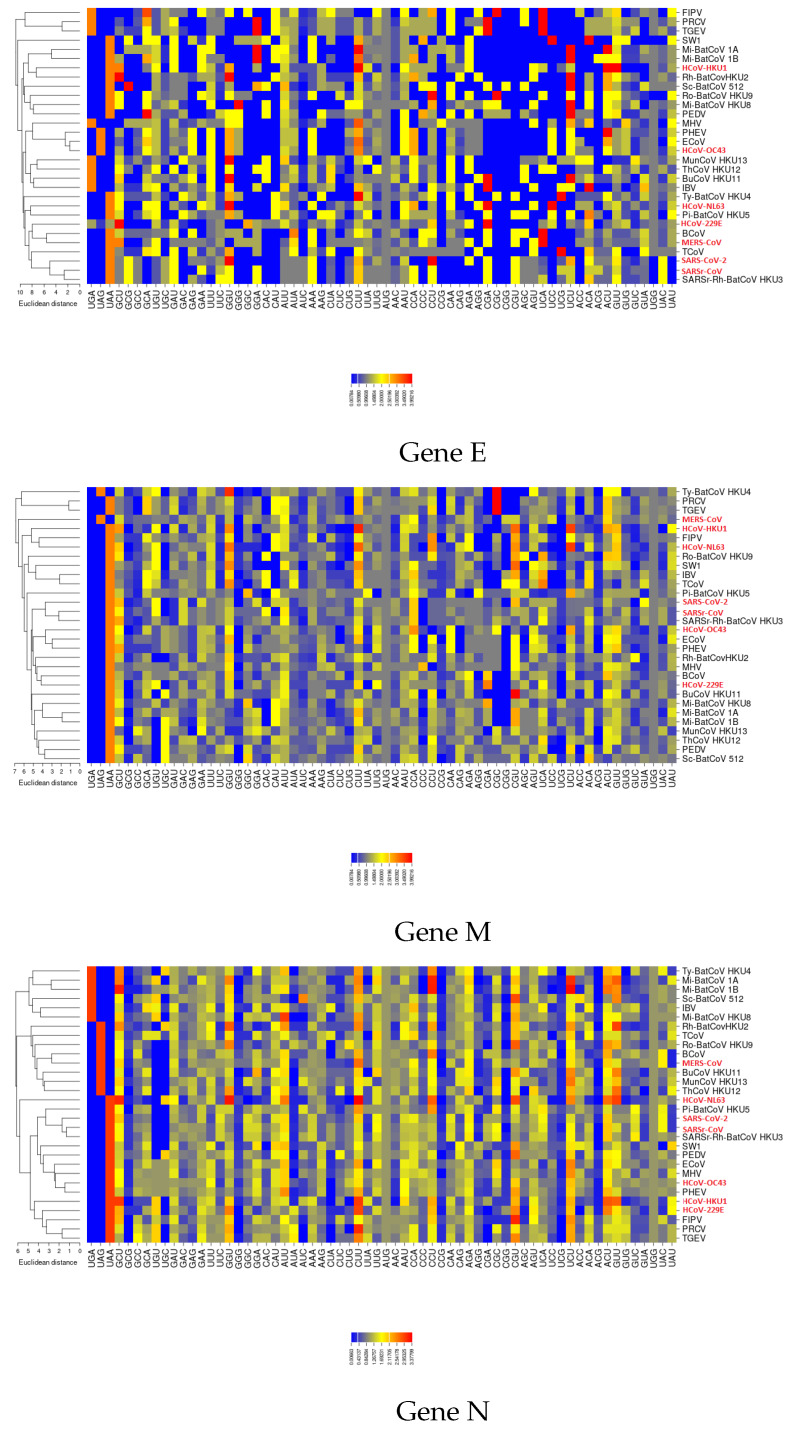
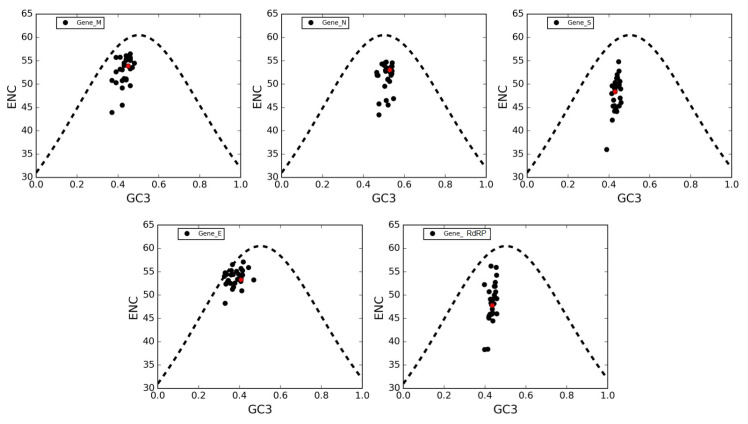
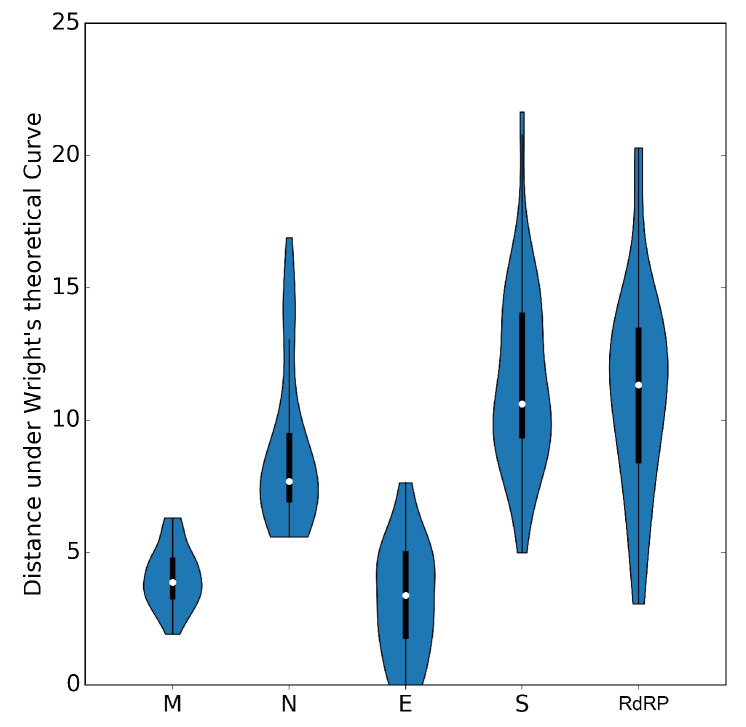
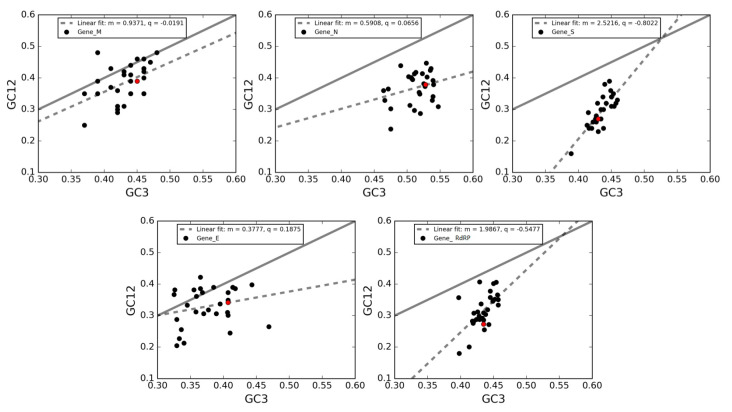
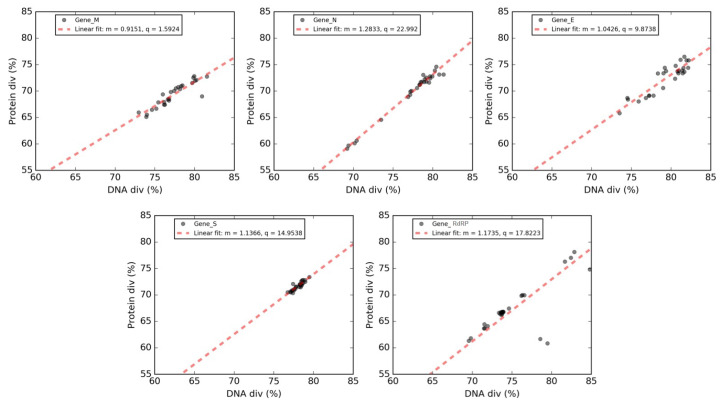
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which first occurred in Wuhan (China) in December of 2019, causes a severe acute respiratory illness with a high mortality rate, and has spread around the world. To gain an understanding of the evolution of the newly emerging SARS-CoV-2, we herein analyzed the codon usage pattern of SARS-CoV-2. For this purpose, we compared the codon usage of SARS-CoV-2 with that of other viruses belonging to the subfamily of Orthocoronavirinae. We found that SARS-CoV-2 has a high AU content that strongly influences its codon usage, which appears to be better adapted to the human host. We also studied the evolutionary pressures that influence the codon usage of five conserved coronavirus genes encoding the viral replicase, spike, envelope, membrane and nucleocapsid proteins. We found different patterns of both mutational bias and natural selection that affect the codon usage of these genes. Moreover, we show here that the two integral membrane proteins (matrix and envelope) tend to evolve slowly by accumulating nucleotide mutations on their corresponding genes. Conversely, genes encoding nucleocapsid (N), viral replicase and spike proteins (S), although they are regarded as are important targets for the development of vaccines and antiviral drugs, tend to evolve faster in comparison to the two genes mentioned above. Overall, our results suggest that the higher divergence observed for the latter three genes could represent a significant barrier in the development of antiviral therapeutics against SARS-CoV-2.
Keywords: SARS-CoV-2; codon usage bias; coronaviruses; host adaptation; mutational bias; natural selection.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Siddell S.G., Ziebuhr J., Snijder E.J. Coronaviruses, Toroviruses, and Arteriviruses. Topley Wilson’s Microbiol. Microb. Infect. 2005 doi: 10.1002/9780470688618.taw0245. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
