

Dysregulated signalling pathways in innate immune cells with cystic fibrosis mutations
- PMID: 32367193
- PMCID: PMC7599191
- DOI: 10.1007/s00018-020-03540-9
Dysregulated signalling pathways in innate immune cells with cystic fibrosis mutations
Abstract
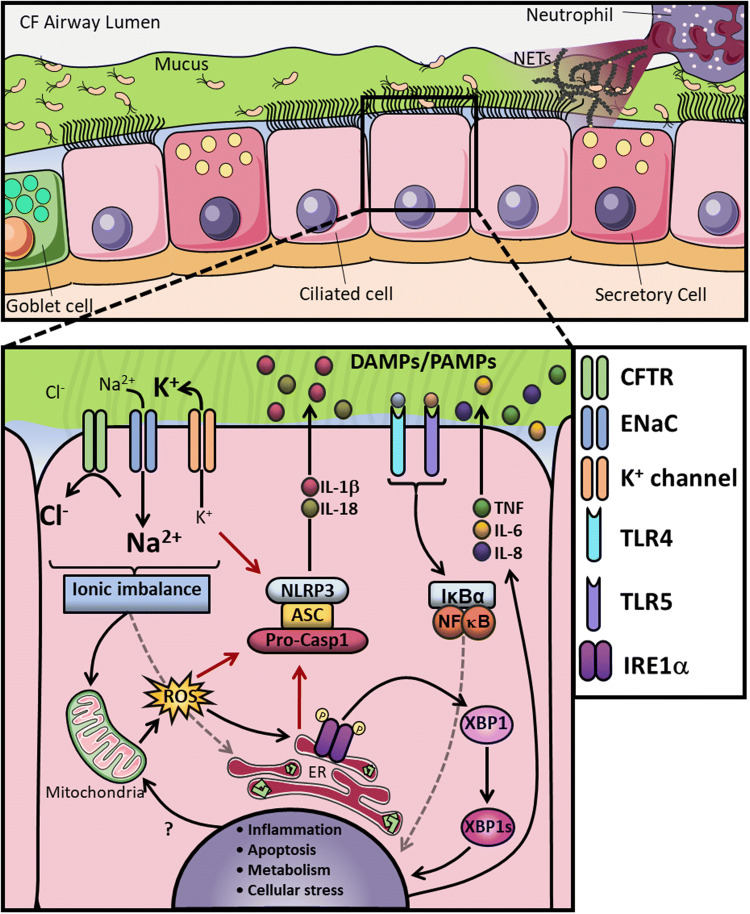
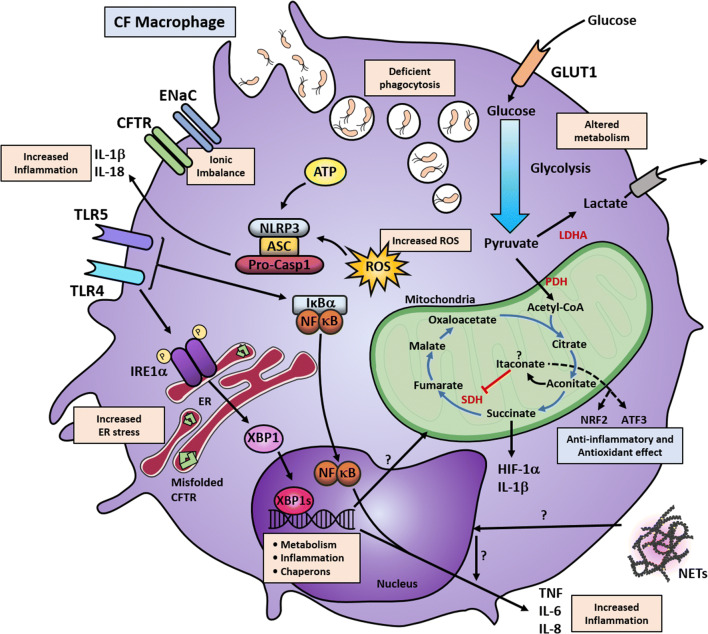
Cystic fibrosis (CF) is one of the most common life-limiting recessive genetic disorders in Caucasians, caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR). CF is a multi-organ disease that involves the lungs, pancreas, sweat glands, digestive and reproductive systems and several other tissues. This debilitating condition is associated with recurrent lower respiratory tract bacterial and viral infections, as well as inflammatory complications that may eventually lead to pulmonary failure. Immune cells play a crucial role in protecting the organs against opportunistic infections and also in the regulation of tissue homeostasis. Innate immune cells are generally affected by CFTR mutations in patients with CF, leading to dysregulation of several cellular signalling pathways that are in continuous use by these cells to elicit a proper immune response. There is substantial evidence to show that airway epithelial cells, neutrophils, monocytes and macrophages all contribute to the pathogenesis of CF, underlying the importance of the CFTR in innate immune responses. The goal of this review is to put into context the important role of the CFTR in different innate immune cells and how CFTR dysfunction contributes to the pathogenesis of CF, highlighting several signalling pathways that may be dysregulated in cells with CFTR mutations.
Keywords: CFTR and autoinflammation; Cystic fibrosis; Inflammation; Macrophages; Monocytes; Neutrophils.
Figures
References
-
- ECFS Patient Registry Annual Data Report (2017) European cystic fibrosis society. https://www.ecfs.eu/sites/default/files/general-content-images/working-g.... Accessed 21 Oct 2019
-
- Patient Registry Annual Data Report (2017) The cystic fibrosis foundation. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-.... Accessed 22 Oct 2019
-
- Stoltz DA, Rokhlina T, Ernst SE, Pezzulo AA, Ostedgaard LS, Karp PH, Samuel MS, Reznikov LR, Rector MV, Gansemer ND, Bouzek DC, Abou Alaiwa MH, Hoegger MJ, Ludwig PS, Taft PJ, Wallen TJ, Wohlford-Lenane C, McMenimen JD, Chen JH, Bogan KL, Adam RJ, Hornick EE, Nelson GAT, Hoffman EA, Chang EH, Zabner J, McCray PB, Jr, Prather RS, Meyerholz DK, Welsh MJ. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123(6):2685–2693. doi: 10.1172/JCI68867. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
