

Periodic DFT Calculations-Review of Applications in the Pharmaceutical Sciences
- PMID: 32369915
- PMCID: PMC7284980
- DOI: 10.3390/pharmaceutics12050415
Periodic DFT Calculations-Review of Applications in the Pharmaceutical Sciences
Abstract
In the introduction to this review the complex chemistry of solid-state pharmaceutical compounds is summarized. It is also explained why the density functional theory (DFT) periodic calculations became recently so popular in studying the solid APIs (active pharmaceutical ingredients). Further, the most popular programs enabling DFT periodic calculations are presented and compared. Subsequently, on the large number of examples, the applications of such calculations in pharmaceutical sciences are discussed. The mentioned topics include, among others, validation of the experimentally obtained crystal structures and crystal structure prediction, insight into crystallization and solvation processes, development of new polymorph synthesis ways, and formulation techniques as well as application of the periodic DFT calculations in the drug analysis.
Keywords: API; CASTEP; DFT; crystal; periodic.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
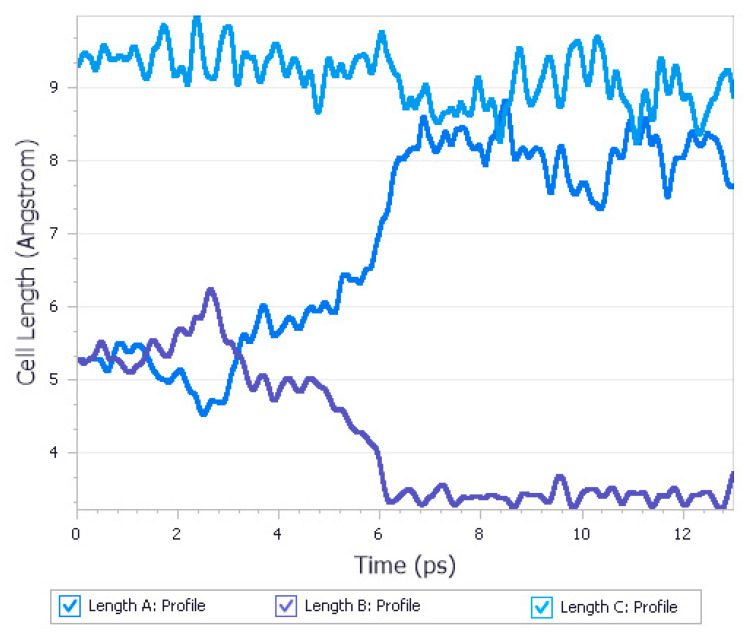
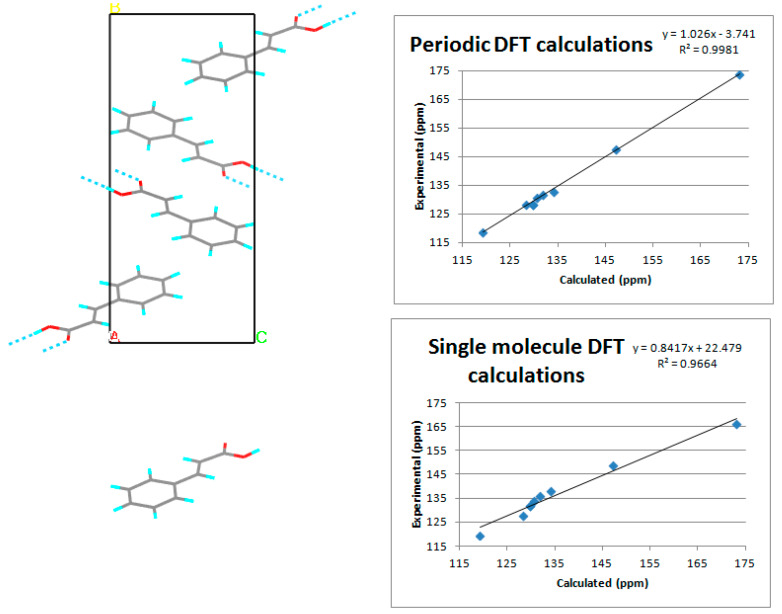
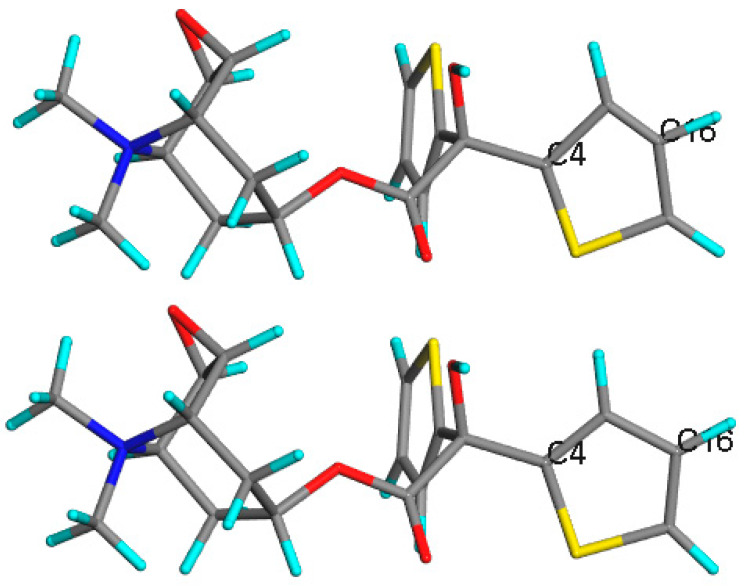
Similar articles
-
Density Functional Theory and Density Functional Tight Binding Studies of Thiamine Hydrochloride Hydrates.Molecules. 2023 Nov 9;28(22):7497. doi: 10.3390/molecules28227497. Molecules. 2023. PMID: 38005219 Free PMC article.
-
Application of Density Functional Theory to Molecular Engineering of Pharmaceutical Formulations.Int J Mol Sci. 2025 Apr 1;26(7):3262. doi: 10.3390/ijms26073262. Int J Mol Sci. 2025. PMID: 40244098 Free PMC article. Review.
-
DFT study of the molecular and crystal structure and vibrational analysis of cisplatin.Spectrochim Acta A Mol Biomol Spectrosc. 2017 Apr 5;176:58-66. doi: 10.1016/j.saa.2017.01.008. Epub 2017 Jan 4. Spectrochim Acta A Mol Biomol Spectrosc. 2017. PMID: 28073067
-
Does the choice of the crystal structure influence the results of the periodic DFT calculations? A case of glycine alpha polymorph GIPAW NMR parameters computations.J Comput Chem. 2018 May 30;39(14):853-861. doi: 10.1002/jcc.25161. Epub 2018 Jan 5. J Comput Chem. 2018. PMID: 29315751
-
Polymorphism and crystallization of active pharmaceutical ingredients (APIs).Curr Med Chem. 2009;16(7):884-905. doi: 10.2174/092986709787549299. Curr Med Chem. 2009. PMID: 19275600 Review.
Cited by
-
Virtual Combinatorial Chemistry and Pharmacological Screening: A Short Guide to Drug Design.Int J Mol Sci. 2022 Jan 30;23(3):1620. doi: 10.3390/ijms23031620. Int J Mol Sci. 2022. PMID: 35163543 Free PMC article. Review.
-
Application of Various Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens.Int J Mol Sci. 2020 Sep 3;21(17):6411. doi: 10.3390/ijms21176411. Int J Mol Sci. 2020. PMID: 32899216 Free PMC article. Review.
-
Can We Predict the Isosymmetric Phase Transition? Application of DFT Calculations to Study the Pressure Induced Transformation of Chlorothiazide.Int J Mol Sci. 2021 Sep 18;22(18):10100. doi: 10.3390/ijms221810100. Int J Mol Sci. 2021. PMID: 34576265 Free PMC article.
-
Crystallographic and DFT study of novel dimethoxybenzene derivatives.BMC Chem. 2025 May 14;19(1):127. doi: 10.1186/s13065-025-01496-0. BMC Chem. 2025. PMID: 40369581 Free PMC article.
-
Density Functional Theory and Density Functional Tight Binding Studies of Thiamine Hydrochloride Hydrates.Molecules. 2023 Nov 9;28(22):7497. doi: 10.3390/molecules28227497. Molecules. 2023. PMID: 38005219 Free PMC article.
References
-
- Ouyang D., Smith S.C. Computational Pharmaceutics: Application of Molecular Modeling in Drug Delivery. John Wiley & Sons; Hoboken, NJ, USA: 2015.
Publication types
LinkOut - more resources
Full Text Sources
