

Chronic mild hypoxia accelerates recovery from preexisting EAE by enhancing vascular integrity and apoptosis of infiltrated monocytes
- PMID: 32371484
- PMCID: PMC7245138
- DOI: 10.1073/pnas.1920935117
Chronic mild hypoxia accelerates recovery from preexisting EAE by enhancing vascular integrity and apoptosis of infiltrated monocytes
Abstract
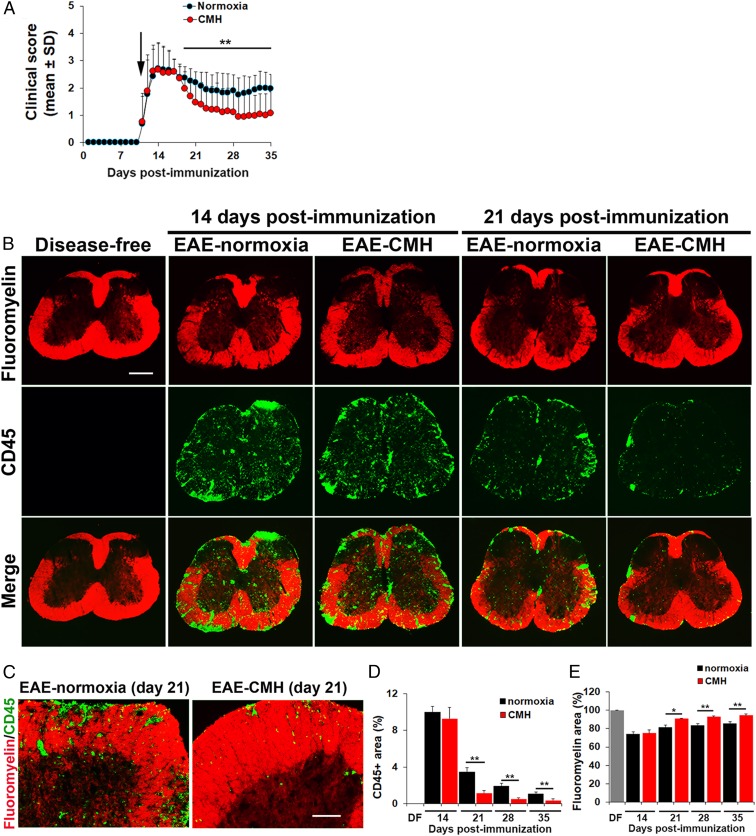
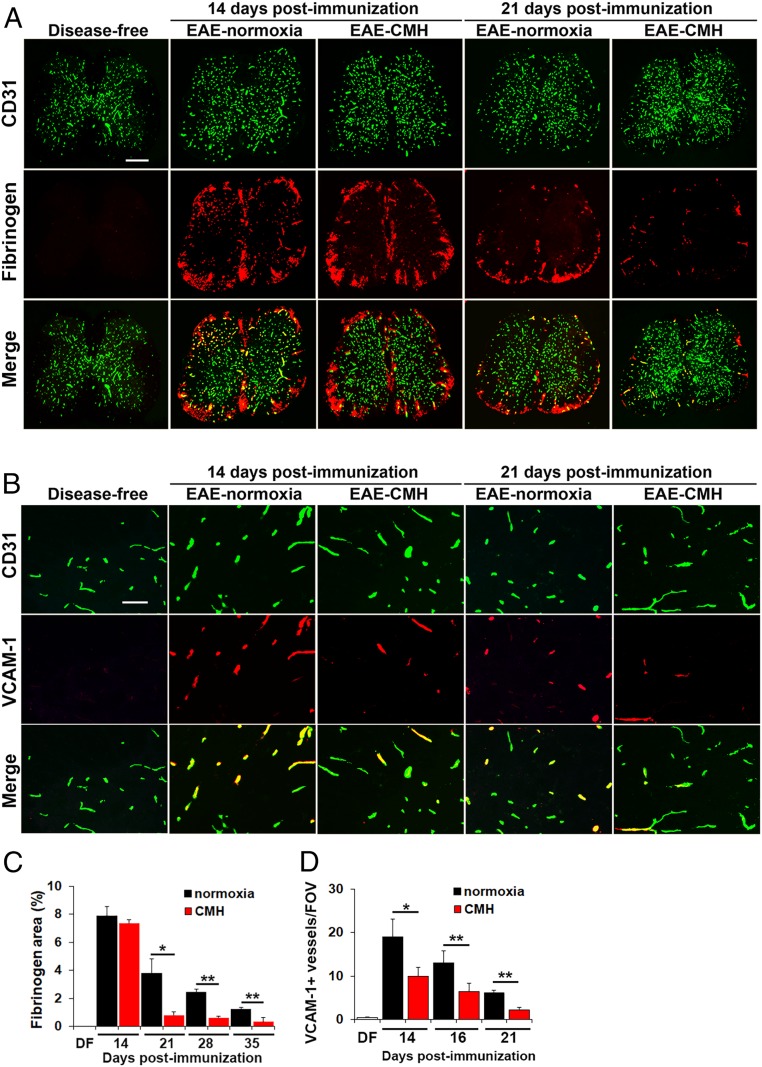
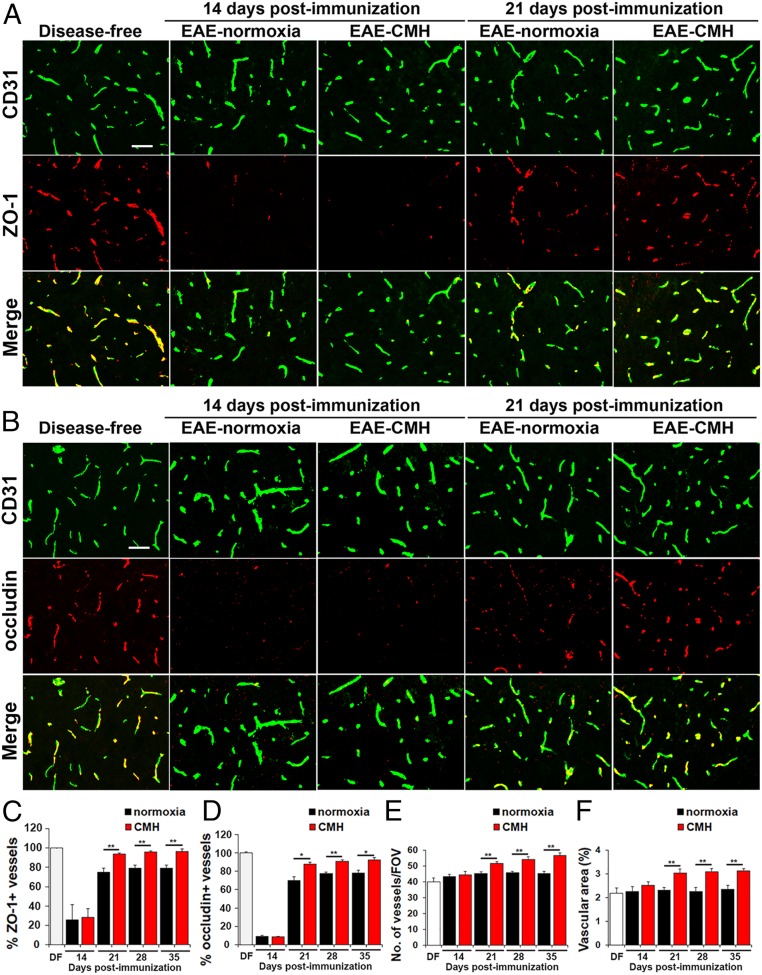
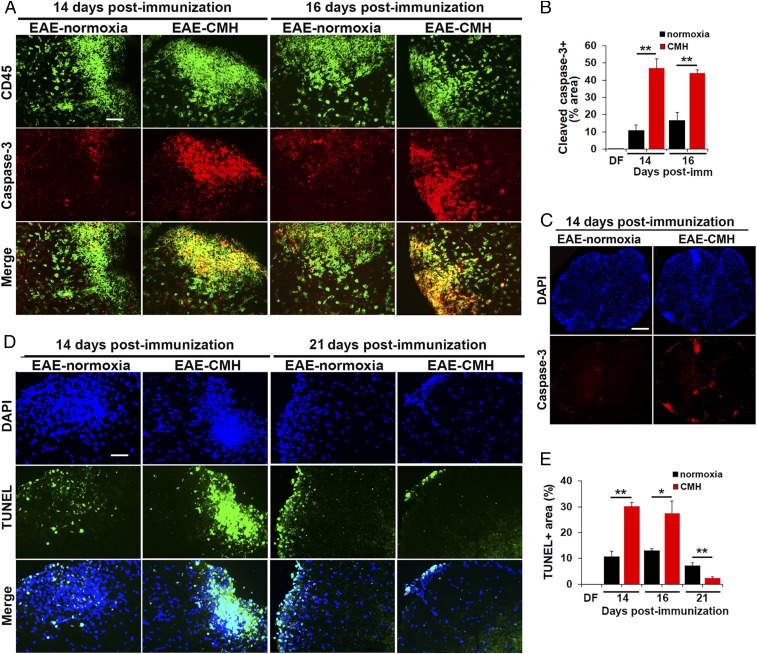
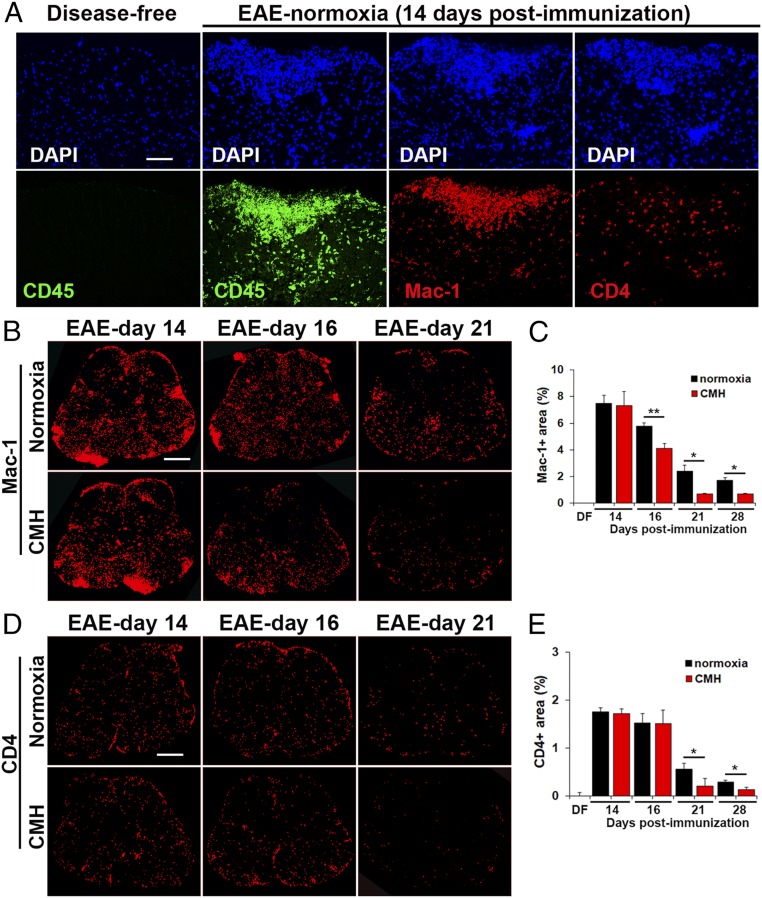
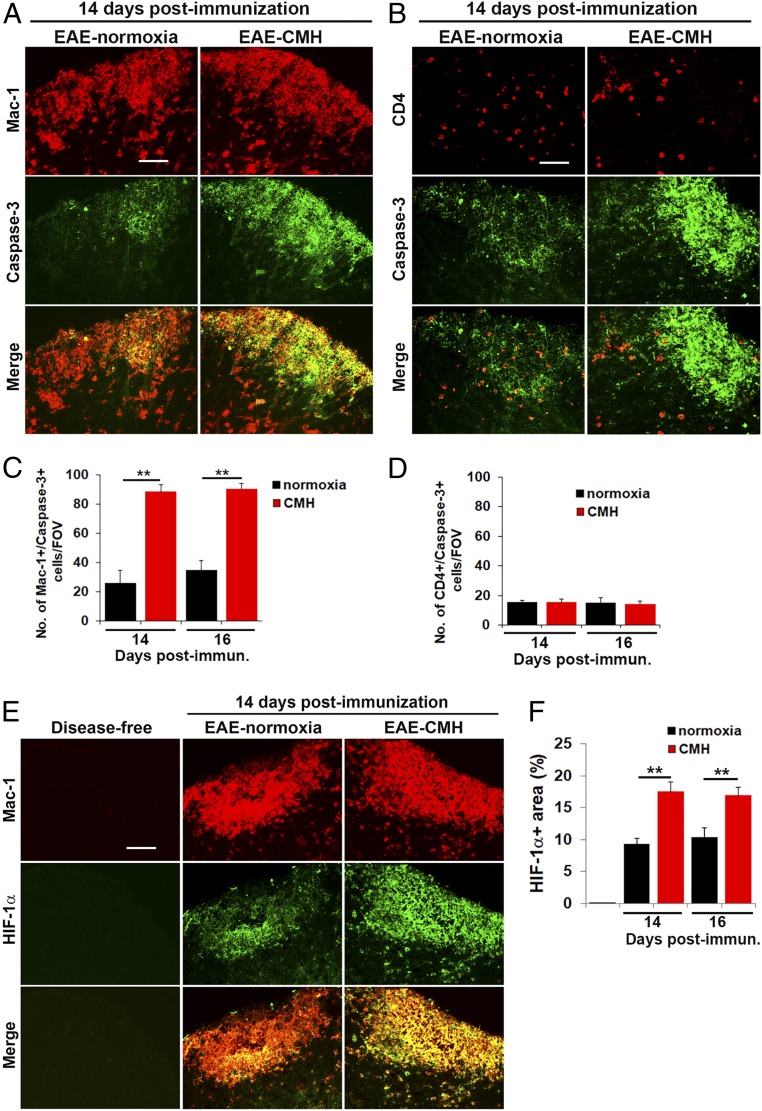
While several studies have shown that hypoxic preconditioning suppresses development of the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS), no one has yet examined the important clinically relevant question of whether mild hypoxia can impact the progression of preexisting disease. Using a relapsing-remitting model of EAE, here we demonstrate that when applied to preexisting disease, chronic mild hypoxia (CMH, 10% O2) markedly accelerates clinical recovery, leading to long-term stable reductions in clinical score. At the histological level, CMH led to significant reductions in vascular disruption, leukocyte accumulation, and demyelination. Spinal cord blood vessels of CMH-treated mice showed reduced expression of the endothelial activation molecule VCAM-1 but increased expression of the endothelial tight junction proteins ZO-1 and occludin, key mechanisms underlying vascular integrity. Interestingly, while equal numbers of inflammatory leukocytes were present in the spinal cord at peak disease (day 14 postimmunization; i.e., 3 d after CMH started), apoptotic removal of infiltrated leukocytes during the remission phase was markedly accelerated in CMH-treated mice, as determined by increased numbers of monocytes positive for TUNEL and cleaved caspase-3. The enhanced monocyte apoptosis in CMH-treated mice was paralleled by increased numbers of HIF-1α+ monocytes, suggesting that CMH enhances monocyte removal by amplifying the hypoxic stress manifest within monocytes in acute inflammatory lesions. These data demonstrate that mild hypoxia promotes recovery from preexisting inflammatory demyelinating disease and suggest that this protection is primarily the result of enhanced vascular integrity and accelerated apoptosis of infiltrated monocytes.
Keywords: blood–brain barrier; chronic mild hypoxia; experimental autoimmune encephalomyelitis; neuroinflammation; vascular integrity.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Wingerchuk D. M., Carter J. L., Multiple sclerosis: Current and emerging disease-modifying therapies and treatment strategies. Mayo Clin. Proc. 89, 225–240 (2014). - PubMed
-
- Ffrench-Constant C., Pathogenesis of multiple sclerosis. Lancet 343, 271–275 (1994). - PubMed
-
- Lassmann H., “Multiple sclerosis pathology” in McAlpine’s Multiple Sclerosis, Compston A., Ed. (Churchill Livingstone, ed. 3, 1998), pp. 323–358.
-
- Dowden J., Corbett D., Ischemic preconditioning in 18- to 20-month-old gerbils: Long-term survival with functional outcome measures. Stroke 30, 1240–1246 (1999). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
