

Long-range Regulation of Partially Folded Amyloidogenic Peptides
- PMID: 32371882
- PMCID: PMC7200734
- DOI: 10.1038/s41598-020-64303-x
Long-range Regulation of Partially Folded Amyloidogenic Peptides
Abstract
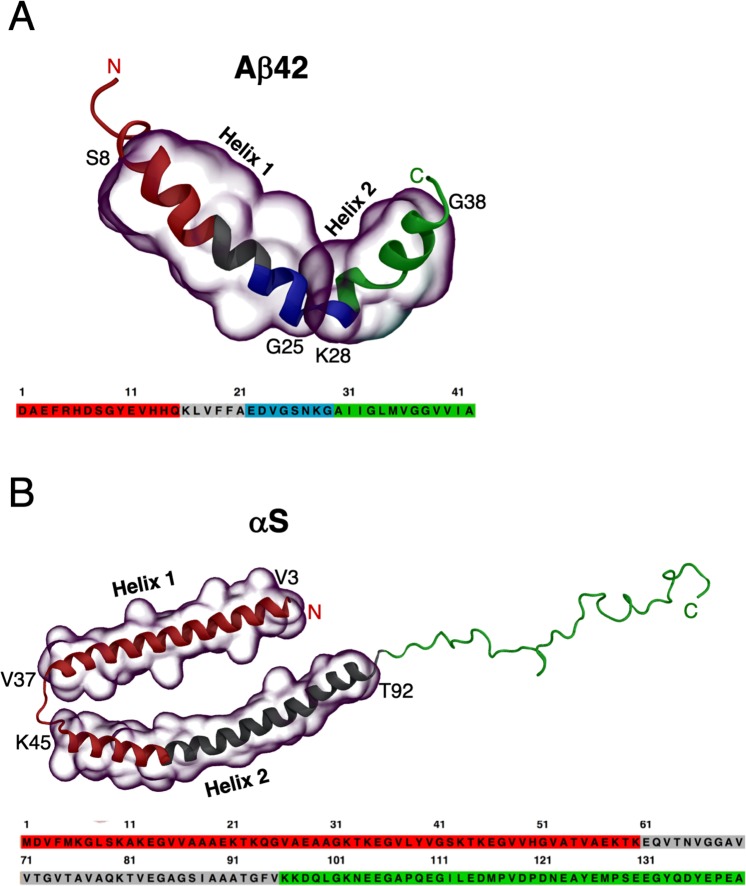
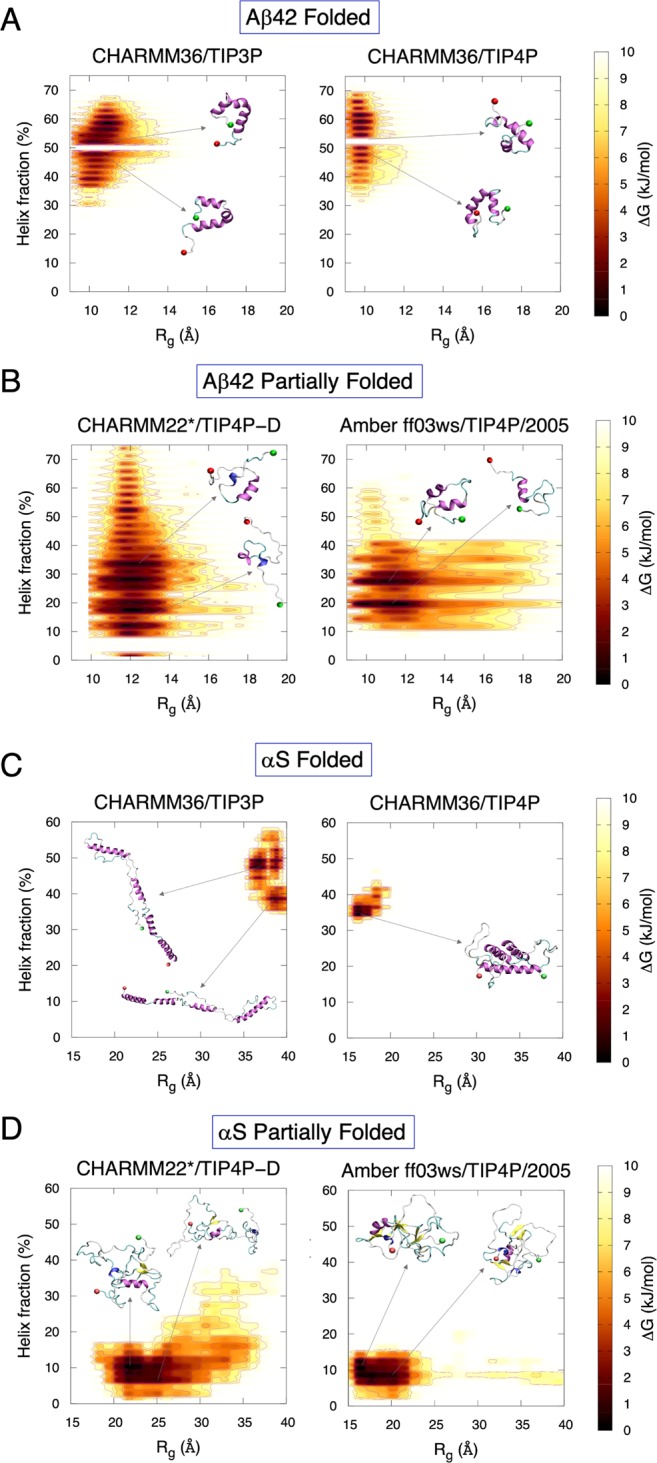
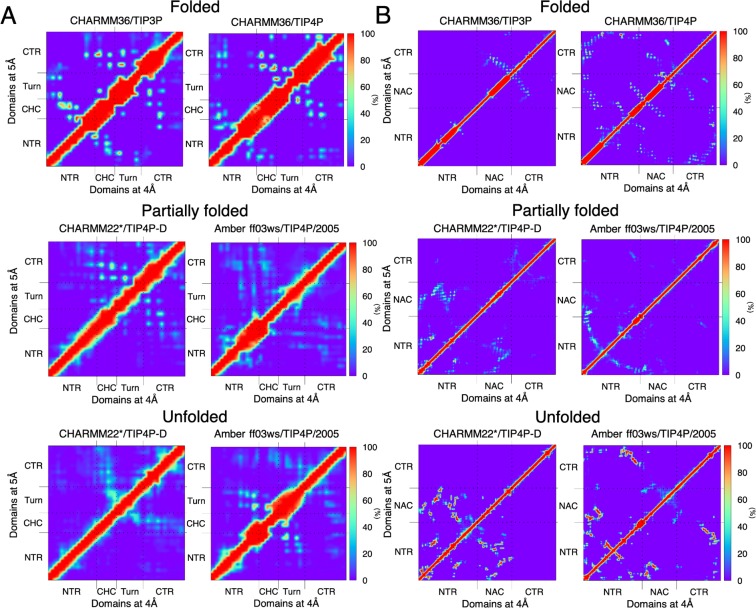
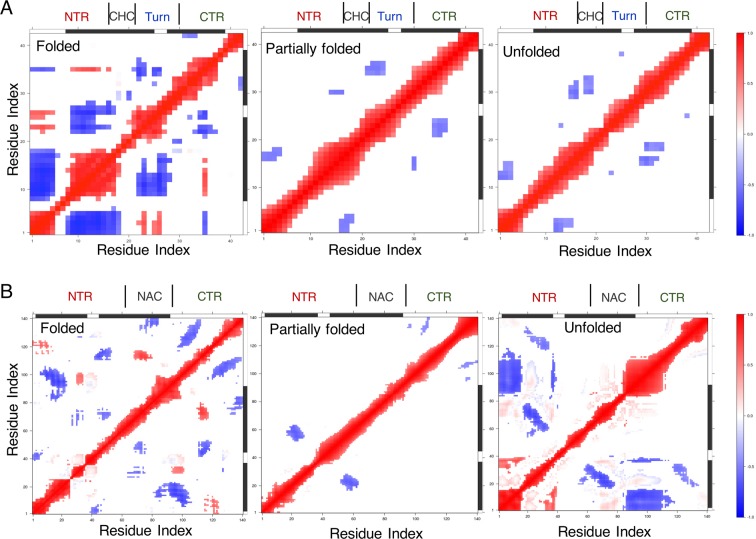
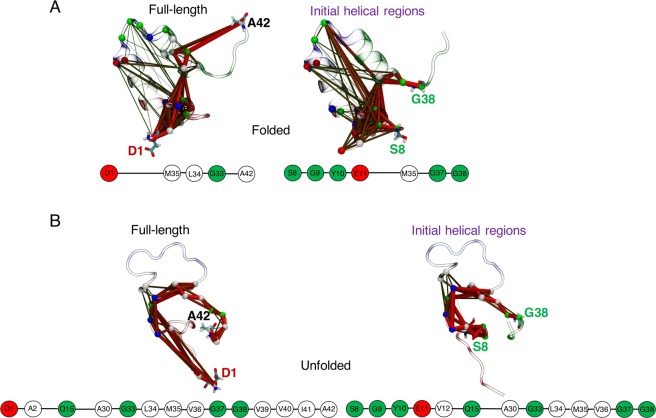
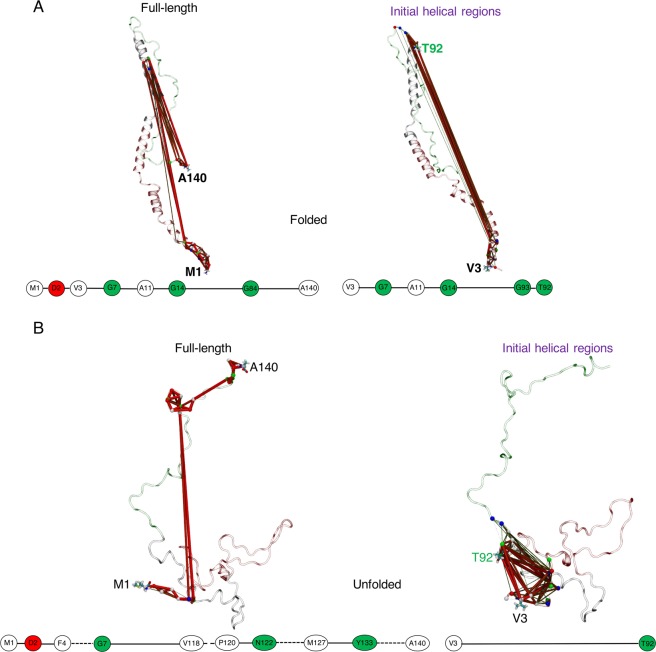
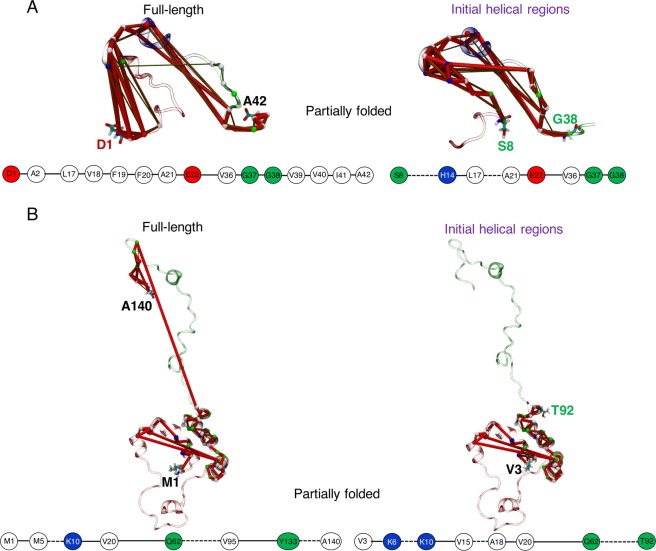
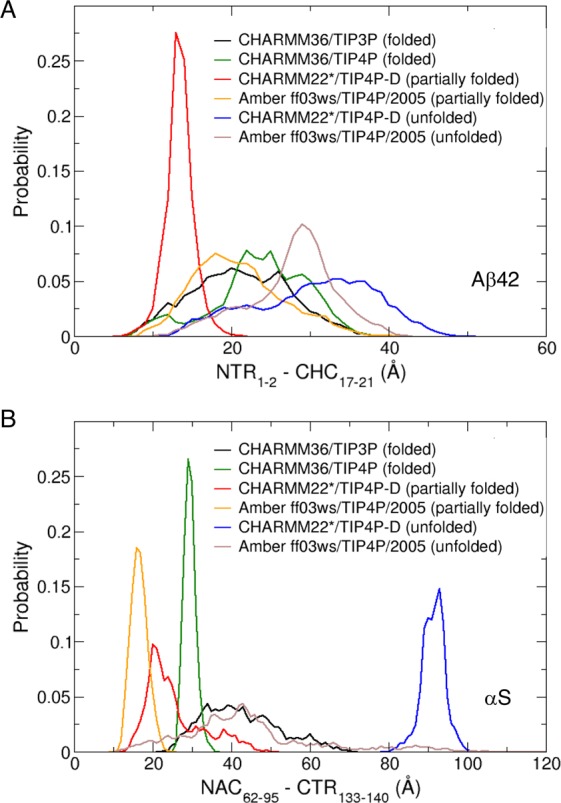
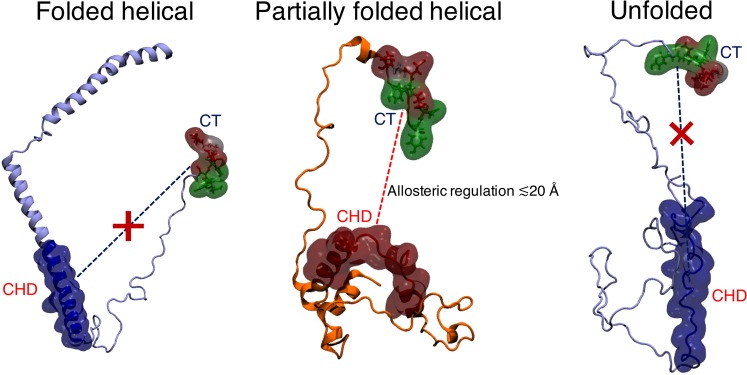
Neurodegeneration involves abnormal aggregation of intrinsically disordered amyloidogenic peptides (IDPs), usually mediated by hydrophobic protein-protein interactions. There is mounting evidence that formation of α-helical intermediates is an early event during self-assembly of amyloid-β42 (Aβ42) and α-synuclein (αS) IDPs in Alzheimer's and Parkinson's disease pathogenesis, respectively. However, the driving force behind on-pathway molecular assembly of partially folded helical monomers into helical oligomers assembly remains unknown. Here, we employ extensive molecular dynamics simulations to sample the helical conformational sub-spaces of monomeric peptides of both Aβ42 and αS. Our computed free energies, population shifts, and dynamic cross-correlation network analyses reveal a common feature of long-range intra-peptide modulation of partial helical folds of the amyloidogenic central hydrophobic domains via concerted coupling with their charged terminal tails (N-terminus of Aβ42 and C-terminus of αS). The absence of such inter-domain fluctuations in both fully helical and completely unfolded (disordered) states suggests that long-range coupling regulates the dynamicity of partially folded helices, in both Aβ42 and αS peptides. The inter-domain coupling suggests a form of intra-molecular allosteric regulation of the aggregation trigger in partially folded helical monomers. This approach could be applied to study the broad range of amyloidogenic peptides, which could provide a new path to curbing pathogenic aggregation of partially folded conformers into oligomers, by inhibition of sites far from the hydrophobic core.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Molecular Simulations Reveal Terminal Group Mediated Stabilization of Helical Conformers in Both Amyloid-β42 and α-Synuclein.ACS Chem Neurosci. 2019 Jun 19;10(6):2830-2842. doi: 10.1021/acschemneuro.9b00053. Epub 2019 Apr 5. ACS Chem Neurosci. 2019. PMID: 30917651
-
Characterization of Amyloidogenic Peptide Aggregability in Helical Subspace.Methods Mol Biol. 2022;2340:401-448. doi: 10.1007/978-1-0716-1546-1_18. Methods Mol Biol. 2022. PMID: 35167084
-
Characterization of the Conformations of Amyloid Beta 42 in Solution That May Mediate Its Initial Hydrophobic Aggregation.J Phys Chem B. 2022 Oct 13;126(40):7916-7933. doi: 10.1021/acs.jpcb.2c04743. Epub 2022 Sep 30. J Phys Chem B. 2022. PMID: 36179370
-
Disordered amyloidogenic peptides may insert into the membrane and assemble into common cyclic structural motifs.Chem Soc Rev. 2014 Oct 7;43(19):6750-64. doi: 10.1039/c3cs60459d. Chem Soc Rev. 2014. PMID: 24566672 Free PMC article. Review.
-
Elucidating the Structures of Amyloid Oligomers with Macrocyclic β-Hairpin Peptides: Insights into Alzheimer's Disease and Other Amyloid Diseases.Acc Chem Res. 2018 Mar 20;51(3):706-718. doi: 10.1021/acs.accounts.7b00554. Epub 2018 Mar 6. Acc Chem Res. 2018. PMID: 29508987 Free PMC article. Review.
Cited by
-
Monomeric a-synuclein (aS) inhibits amyloidogenesis of human prion protein (hPrP) by forming a stable aS-hPrP hetero-dimer.Prion. 2021 Dec;15(1):37-43. doi: 10.1080/19336896.2021.1910176. Prion. 2021. PMID: 33849375 Free PMC article.
-
Single-Particle Resolution of Copper-Associated Annular α-Synuclein Oligomers Reveals Potential Therapeutic Targets of Neurodegeneration.ACS Chem Neurosci. 2022 May 4;13(9):1410-1421. doi: 10.1021/acschemneuro.2c00021. Epub 2022 Apr 12. ACS Chem Neurosci. 2022. PMID: 35414168 Free PMC article.
-
Conformational Selection of α-Synuclein Tetramers at Biological Interfaces.J Chem Inf Model. 2024 Oct 28;64(20):8010-8023. doi: 10.1021/acs.jcim.4c01459. Epub 2024 Oct 8. J Chem Inf Model. 2024. PMID: 39377660 Free PMC article.
-
Challenges in Discovering Drugs That Target the Protein-Protein Interactions of Disordered Proteins.Int J Mol Sci. 2022 Jan 28;23(3):1550. doi: 10.3390/ijms23031550. Int J Mol Sci. 2022. PMID: 35163473 Free PMC article. Review.
-
Effect of an Amyloidogenic SARS-COV-2 Protein Fragment on α-Synuclein Monomers and Fibrils.J Phys Chem B. 2022 May 26;126(20):3648-3658. doi: 10.1021/acs.jpcb.2c01254. Epub 2022 May 17. J Phys Chem B. 2022. PMID: 35580331 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
