

Combining native and 'omics' mass spectrometry to identify endogenous ligands bound to membrane proteins
- PMID: 32371966
- PMCID: PMC7332344
- DOI: 10.1038/s41592-020-0821-0
Combining native and 'omics' mass spectrometry to identify endogenous ligands bound to membrane proteins
Abstract
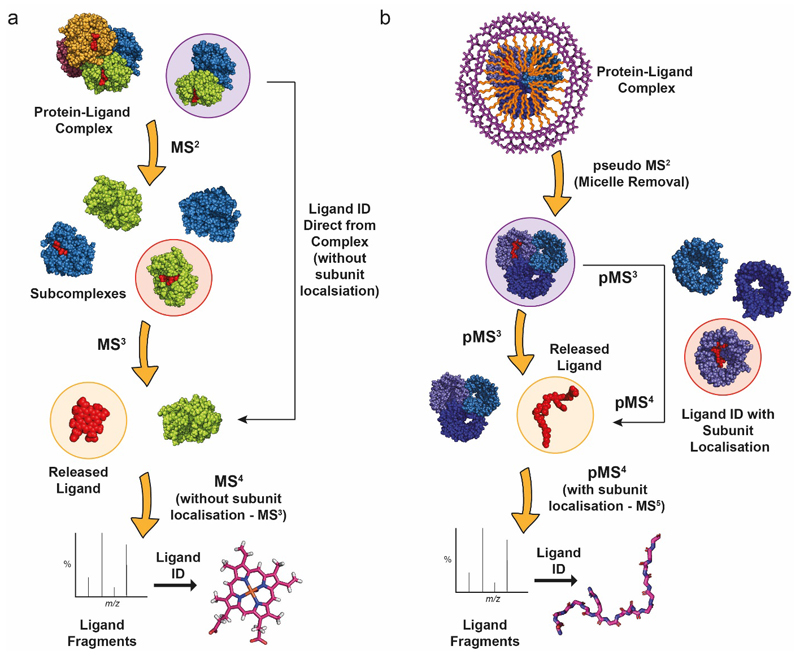
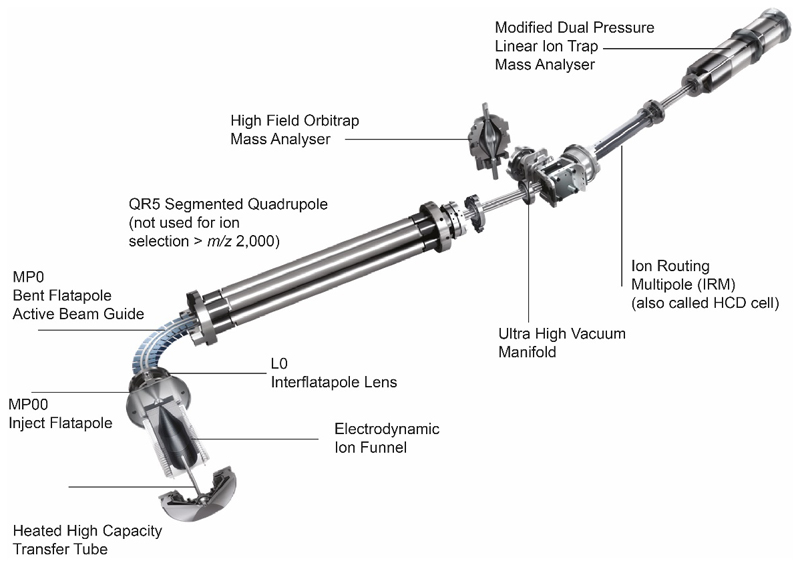
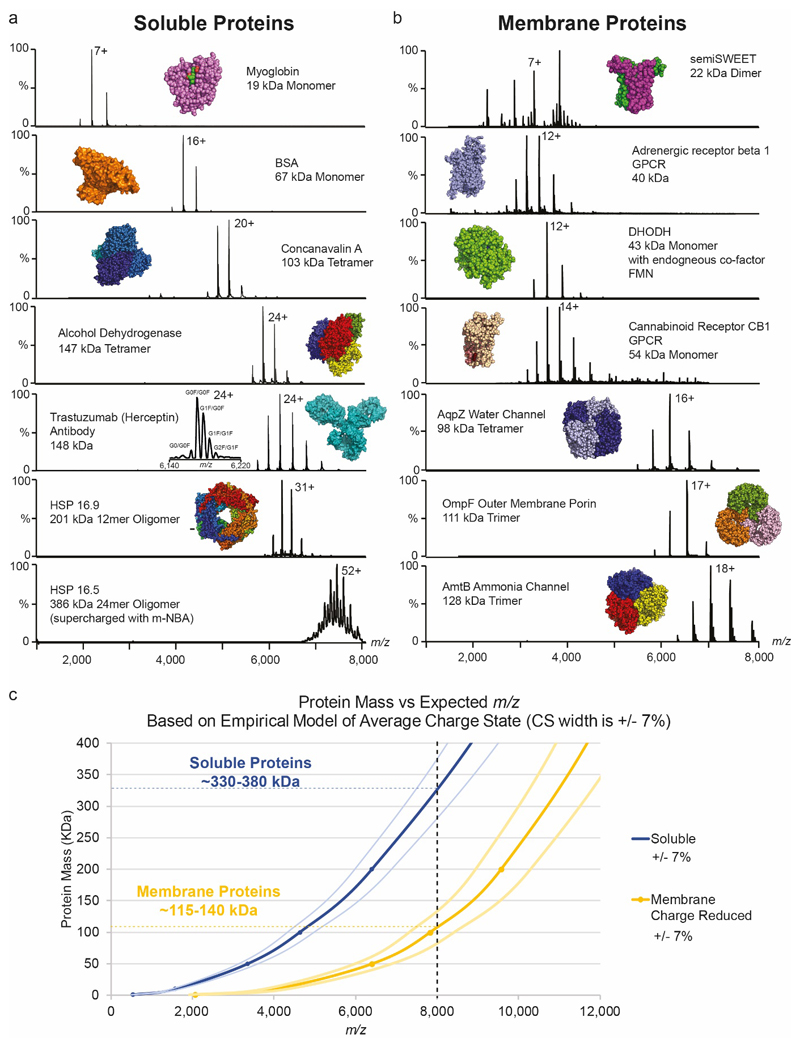
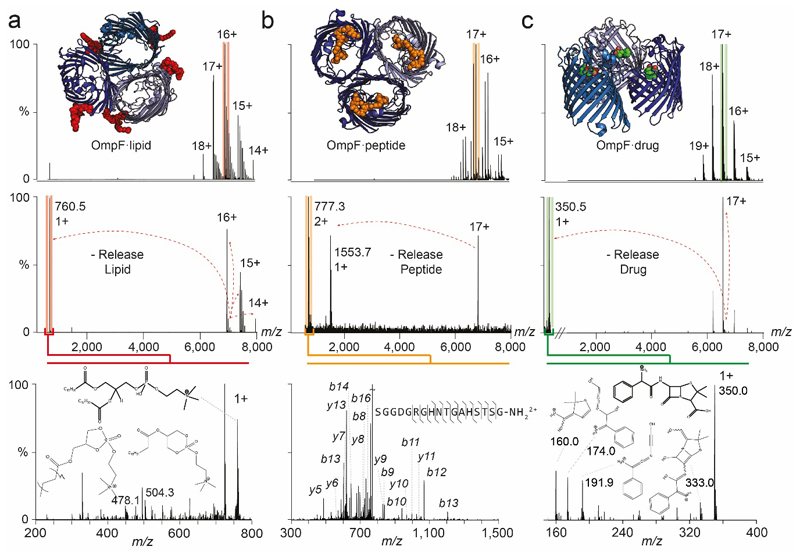
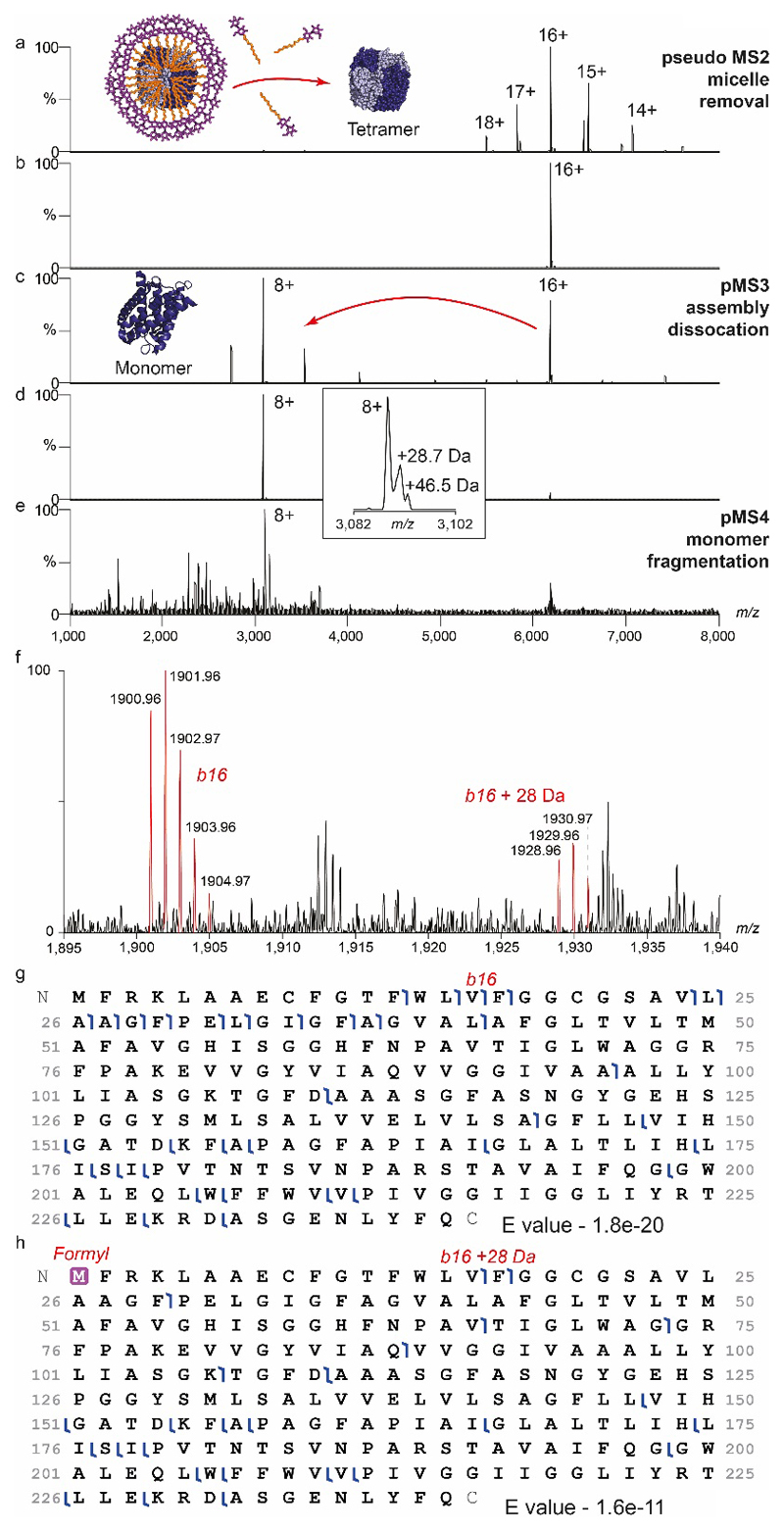
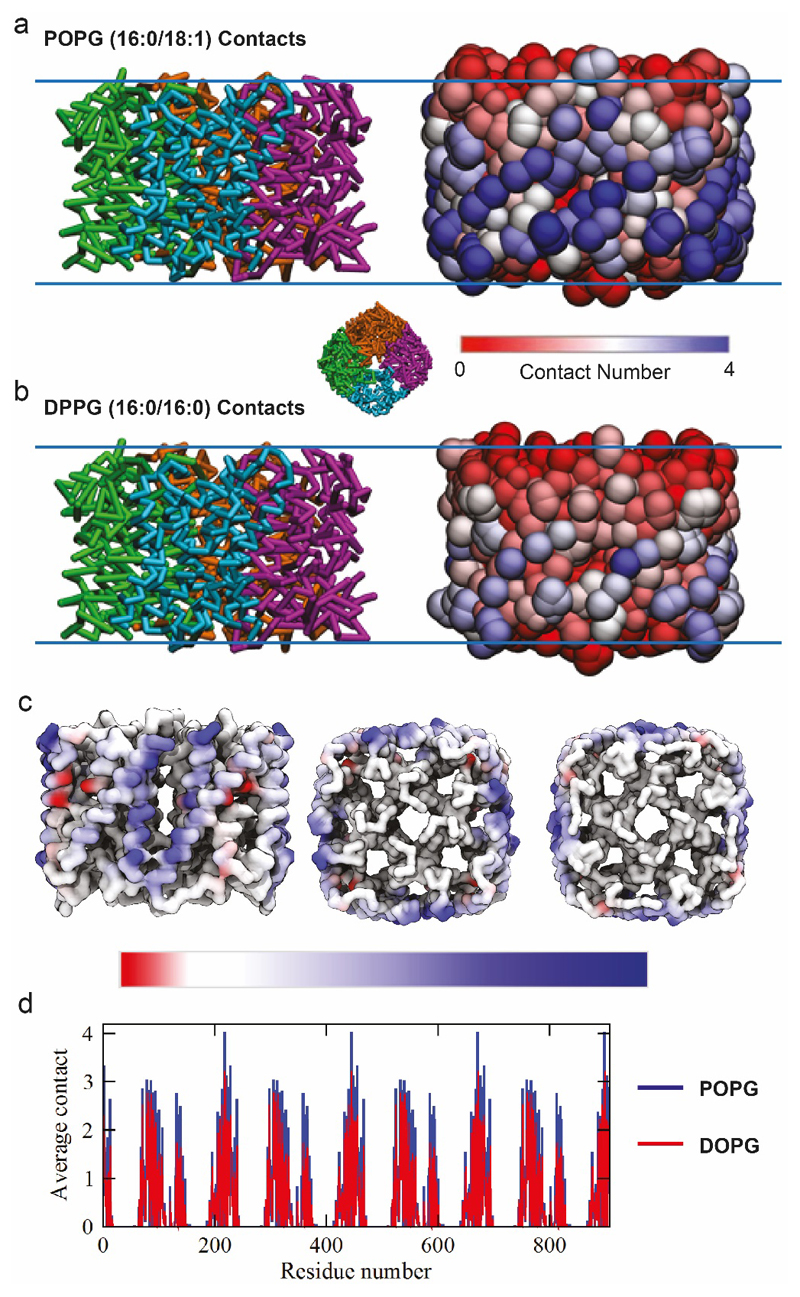
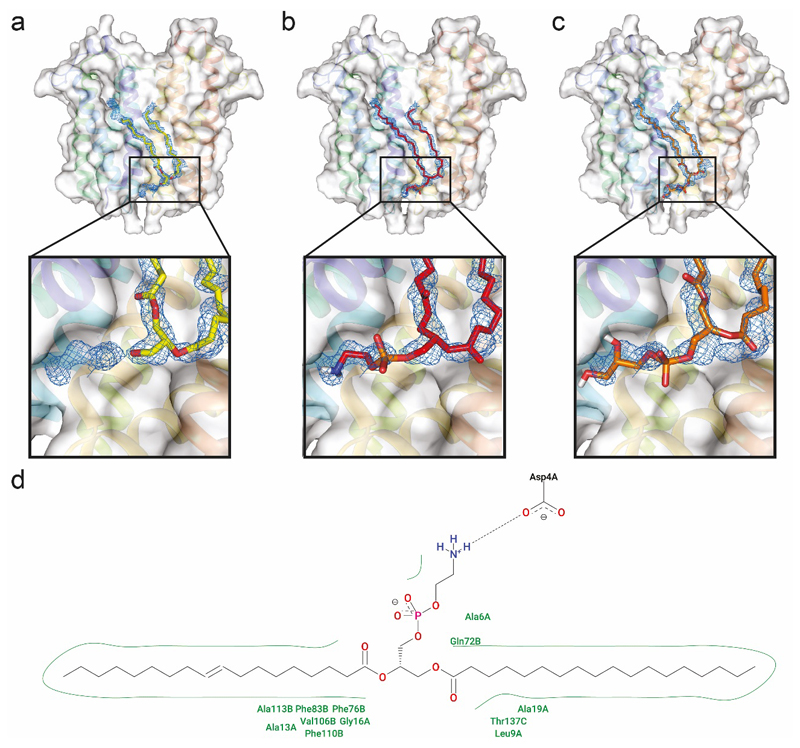
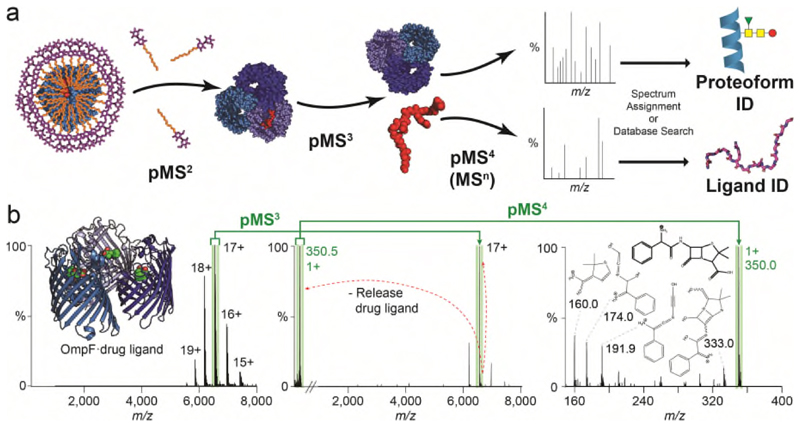
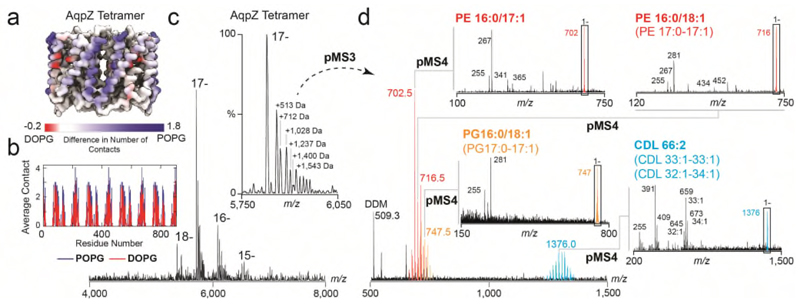
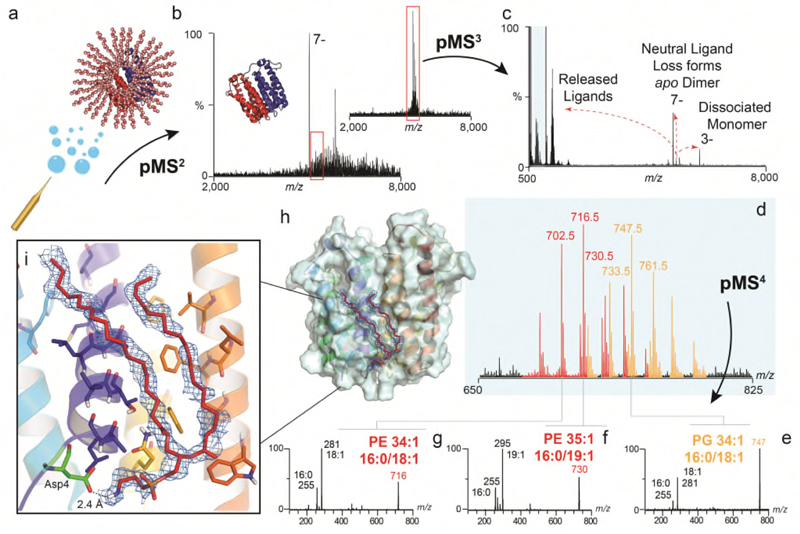
Ligands bound to protein assemblies provide critical information for function, yet are often difficult to capture and define. Here we develop a top-down method, 'nativeomics', unifying 'omics' (lipidomics, proteomics, metabolomics) analysis with native mass spectrometry to identify ligands bound to membrane protein assemblies. By maintaining the link between proteins and ligands, we define the lipidome/metabolome in contact with membrane porins and a mitochondrial translocator to discover potential regulators of protein function.
Conflict of interest statement
IL and HYY are employees of OMass Therapeutics. JG and CVR provide consultancy services to OMass Therapeutics. CM, PR, RH, GM, MG, RV, JS are employees of Thermo Fisher Scientific
Figures
References
-
- Laschet C, Dupuis N, Hanson J. The G protein-coupled receptors deorphanization landscape. Biochem Pharmacol. 2018;153:62–74. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
