

Disentangling the Amyloid Pathways: A Mechanistic Approach to Etiology
- PMID: 32372895
- PMCID: PMC7186396
- DOI: 10.3389/fnins.2020.00256
Disentangling the Amyloid Pathways: A Mechanistic Approach to Etiology
Abstract
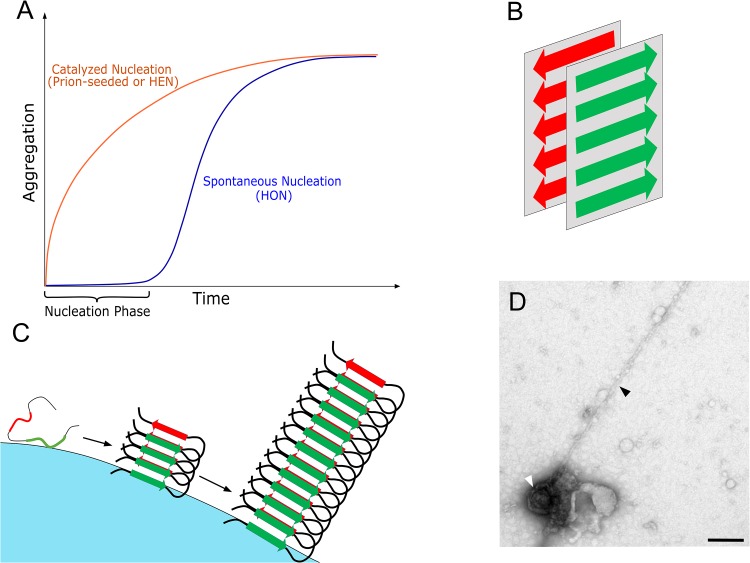
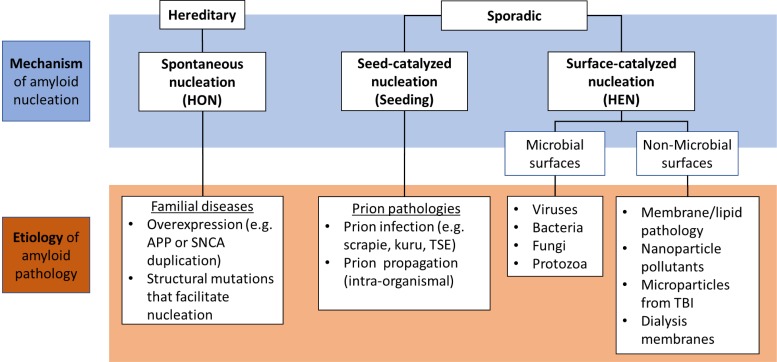
Amyloids are fibrillar protein aggregates associated with diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), type II diabetes and Creutzfeldt-Jakob disease. The process of amyloid polymerization involves three pathological protein transformations; from natively folded conformation to the cross-β conformation, from biophysically soluble to insoluble, and from biologically functional to non-functional. While amyloids share a similar cross-β conformation, the biophysical transformation can either take place spontaneously via a homogeneous nucleation mechanism (HON) or catalytically on an exogenous surface via a heterogeneous nucleation mechanism (HEN). Here, we postulate that the different nucleation pathways can serve as a mechanistic basis for an etiological classification of amyloidopathies, where hereditary forms generally follow the HON pathway, while sporadic forms follow seed-induced (prions) or surface-induced (including microbially induced) HEN pathways. Critically, the conformational and biophysical amyloid transformation results in loss-of-function (LOF) of the original natively folded and soluble protein. This LOF can, at least initially, be the mechanism of amyloid toxicity even before amyloid accumulation reaches toxic levels. By highlighting the important role of non-protein species in amyloid formation and LOF mechanisms of toxicity, we propose a generalized mechanistic framework that could help better understand the diverse etiology of amyloid diseases and offer new opportunities for therapeutic interventions, including replacement therapies.
Keywords: Alzheiemr’s; Parkinson’s; amyloid; nucleation; prion; protein-only hypothesis; virus.
Copyright © 2020 Malmberg, Malm, Gustafsson, Sturchio, Graff, Espay, Wright, El Andaloussi, Lindén and Ezzat.
Figures
Similar articles
-
Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies.Biology (Basel). 2022 Mar 30;11(4):535. doi: 10.3390/biology11040535. Biology (Basel). 2022. PMID: 35453734 Free PMC article. Review.
-
The allure and pitfalls of the prion-like aggregation in neurodegeneration.Handb Clin Neurol. 2023;193:17-22. doi: 10.1016/B978-0-323-85555-6.00004-7. Handb Clin Neurol. 2023. PMID: 36803809 Review.
-
Prion, prionoids and infectious amyloid.Parkinsonism Relat Disord. 2014 Jan;20 Suppl 1:S80-4. doi: 10.1016/S1353-8020(13)70021-X. Parkinsonism Relat Disord. 2014. PMID: 24262195 Review.
-
Key Points Concerning Amyloid Infectivity and Prion-Like Neuronal Invasion.Front Mol Neurosci. 2016 Apr 22;9:29. doi: 10.3389/fnmol.2016.00029. eCollection 2016. Front Mol Neurosci. 2016. PMID: 27147962 Free PMC article.
-
Soluble Amyloid-β Consumption in Alzheimer's Disease.J Alzheimers Dis. 2021;82(4):1403-1415. doi: 10.3233/JAD-210415. J Alzheimers Dis. 2021. PMID: 34151810
Cited by
-
Recalibrating the Why and Whom of Animal Models in Parkinson Disease: A Clinician's Perspective.Brain Sci. 2024 Jan 31;14(2):151. doi: 10.3390/brainsci14020151. Brain Sci. 2024. PMID: 38391726 Free PMC article. Review.
-
Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies.Biology (Basel). 2022 Mar 30;11(4):535. doi: 10.3390/biology11040535. Biology (Basel). 2022. PMID: 35453734 Free PMC article. Review.
-
Microglial amyloid beta clearance is driven by PIEZO1 channels.J Neuroinflammation. 2022 Jun 15;19(1):147. doi: 10.1186/s12974-022-02486-y. J Neuroinflammation. 2022. PMID: 35706029 Free PMC article.
-
High cerebrospinal amyloid-β 42 is associated with normal cognition in individuals with brain amyloidosis.EClinicalMedicine. 2021 Jun 28;38:100988. doi: 10.1016/j.eclinm.2021.100988. eCollection 2021 Aug. EClinicalMedicine. 2021. PMID: 34505023 Free PMC article.
-
The Promising Role of Selenium and Yeast in the Fight Against Protein Amyloidosis.Biol Trace Elem Res. 2025 Mar;203(3):1251-1268. doi: 10.1007/s12011-024-04245-x. Epub 2024 Jun 3. Biol Trace Elem Res. 2025. PMID: 38829477 Free PMC article. Review.
References
-
- Abeliovich A., Schmitz Y., Fariñ I., Choi-Lundberg D., Ho W.-H., Castillo P. E., et al. (2000). Mice Lacking -Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 25 239–252. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources