

Branched chain amino acids exacerbate myocardial ischemia/reperfusion vulnerability via enhancing GCN2/ATF6/PPAR-α pathway-dependent fatty acid oxidation
- PMID: 32373236
- PMCID: PMC7196282
- DOI: 10.7150/thno.44836
Branched chain amino acids exacerbate myocardial ischemia/reperfusion vulnerability via enhancing GCN2/ATF6/PPAR-α pathway-dependent fatty acid oxidation
Abstract
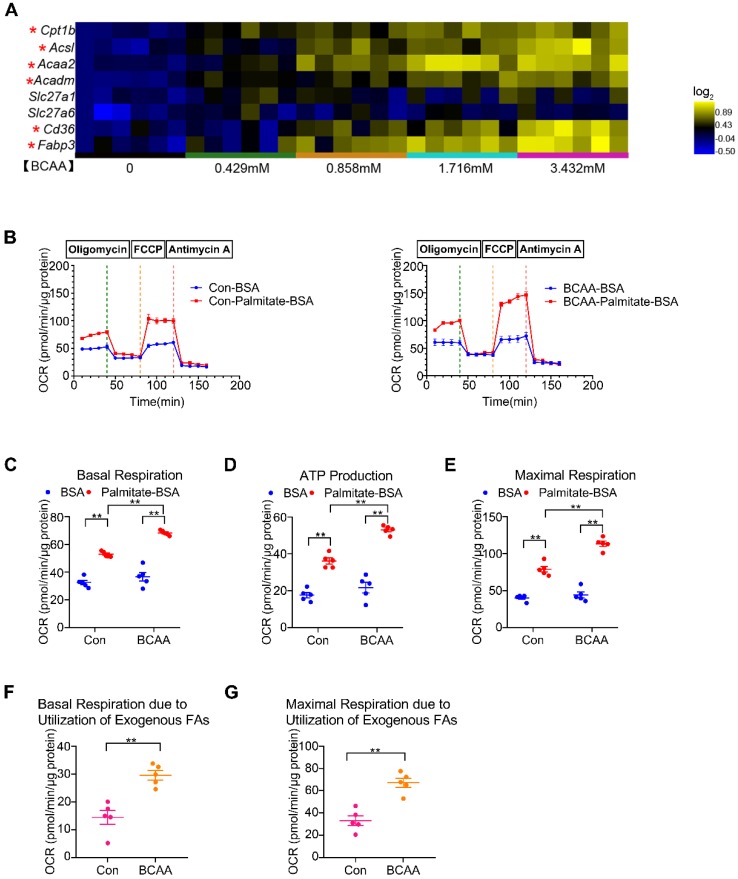
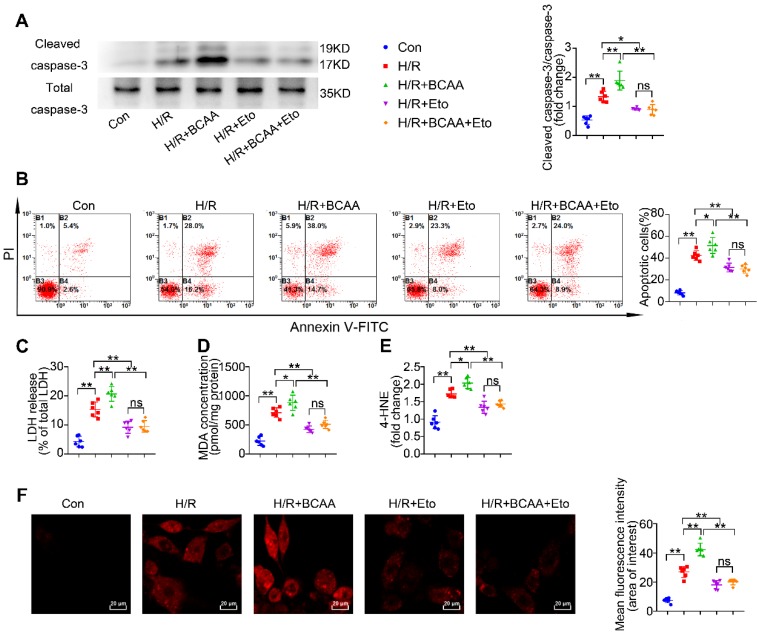
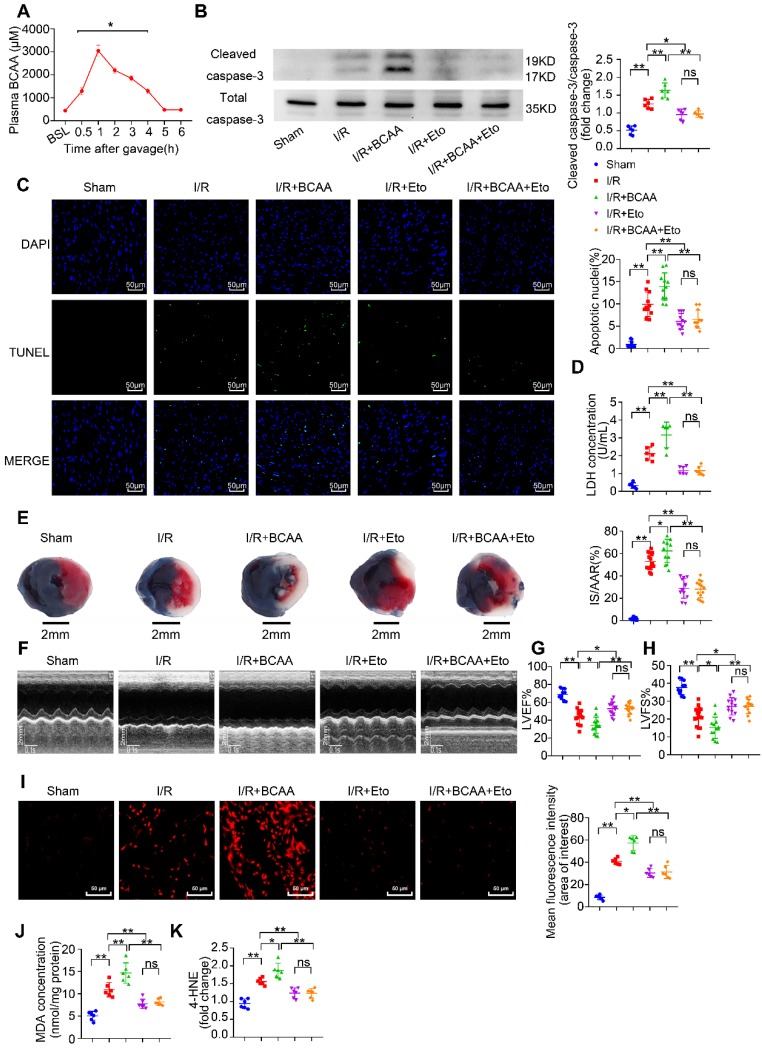
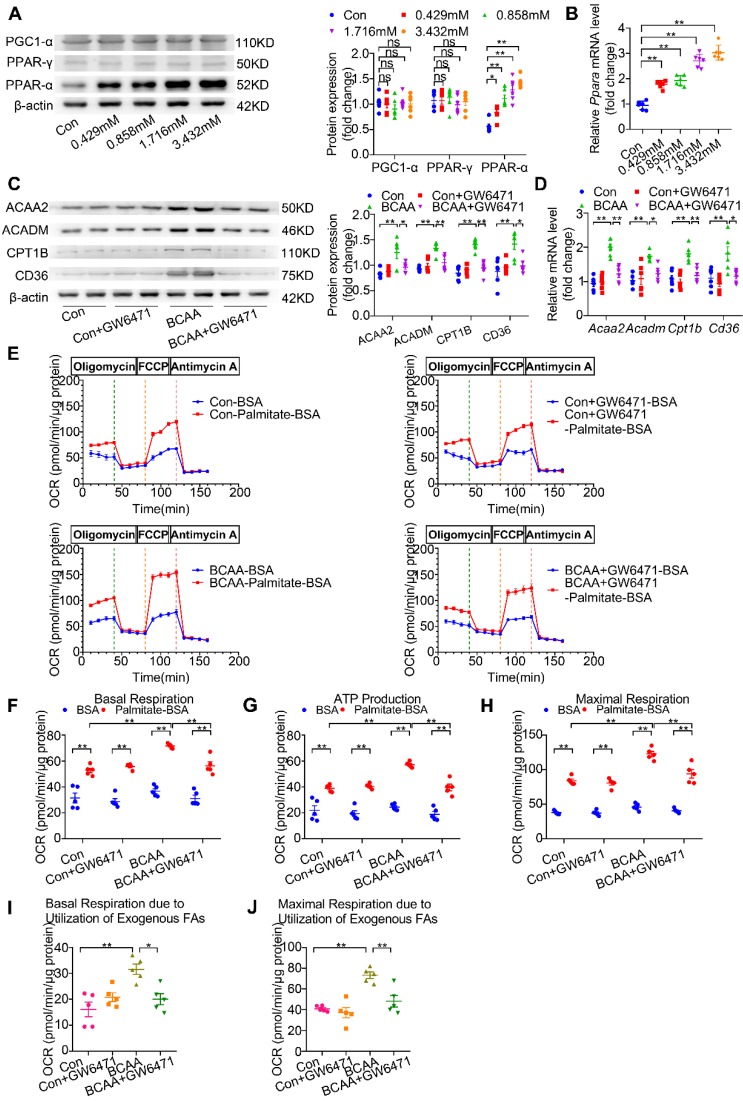
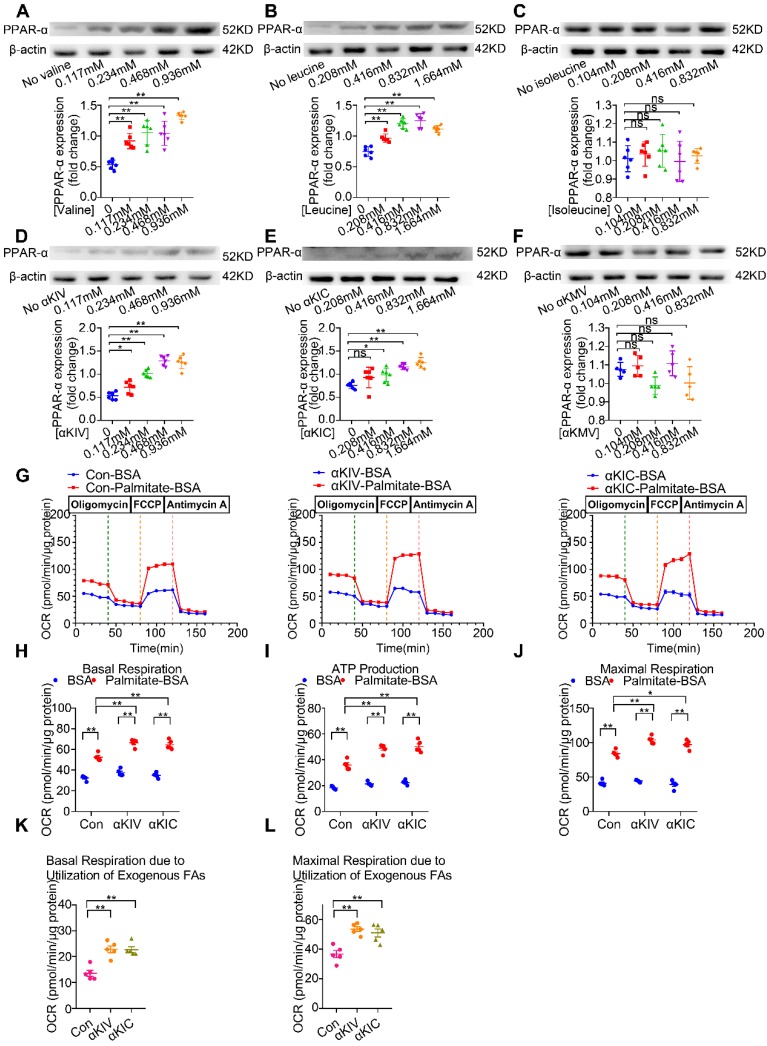
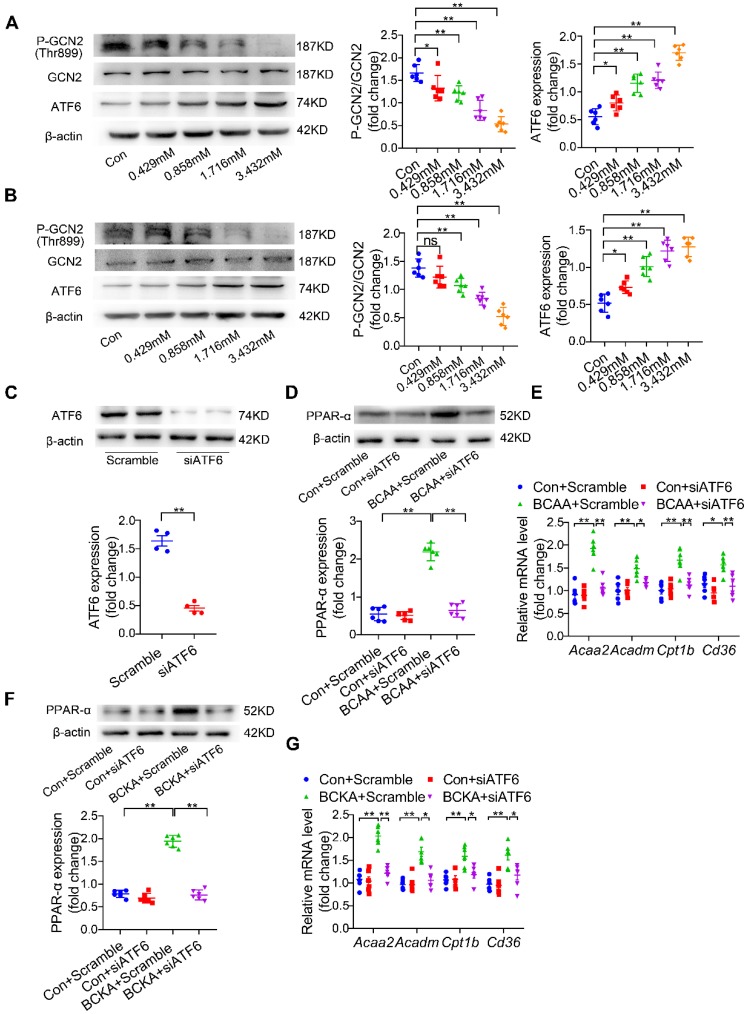
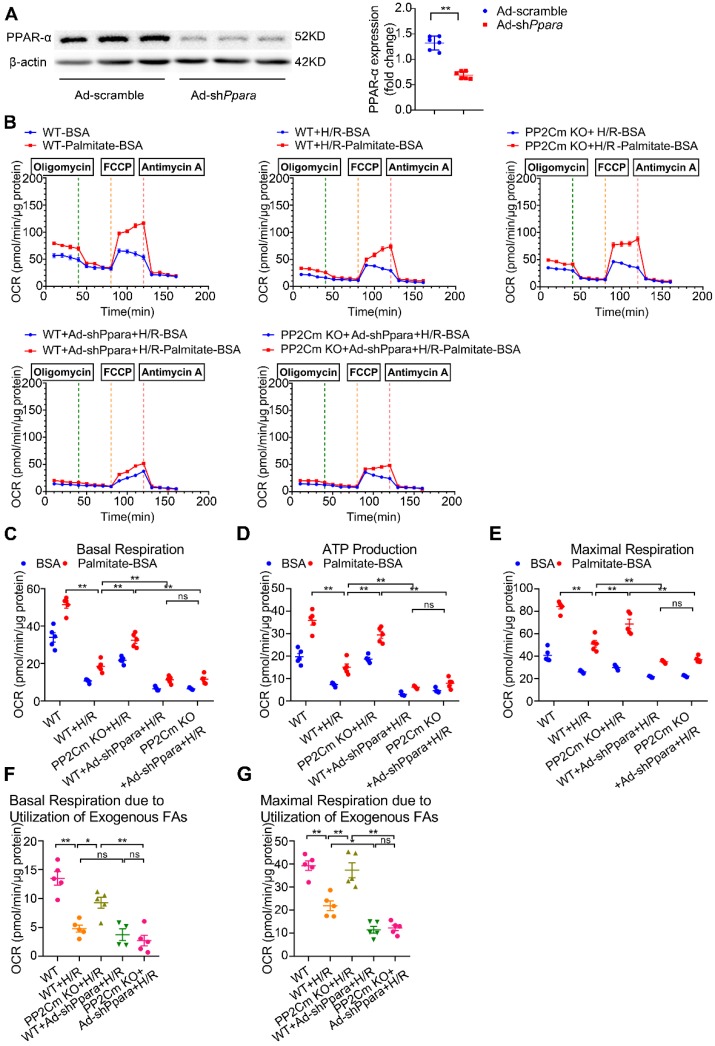
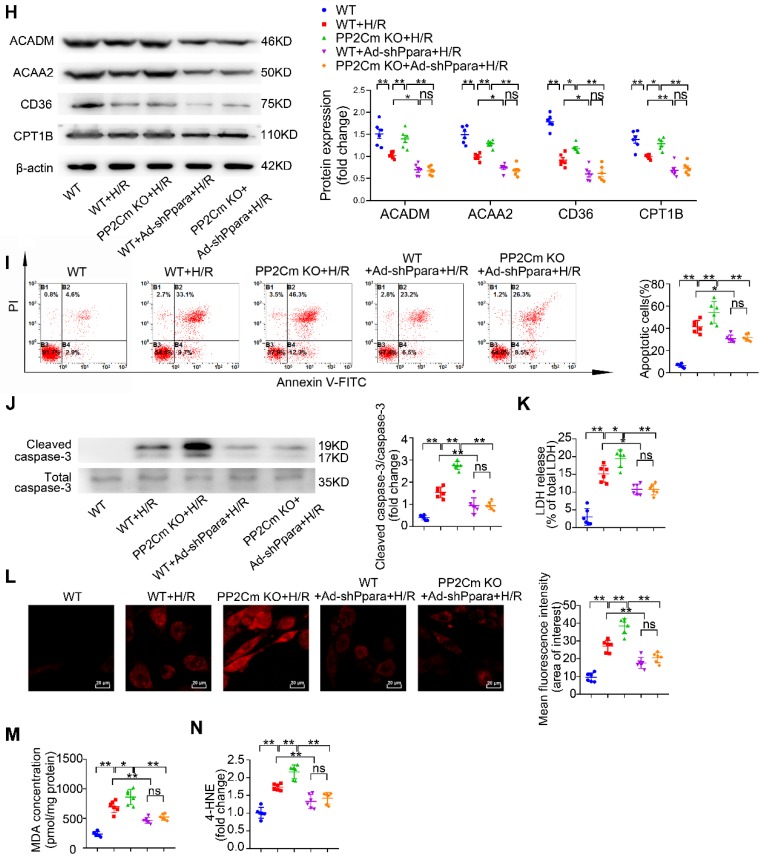
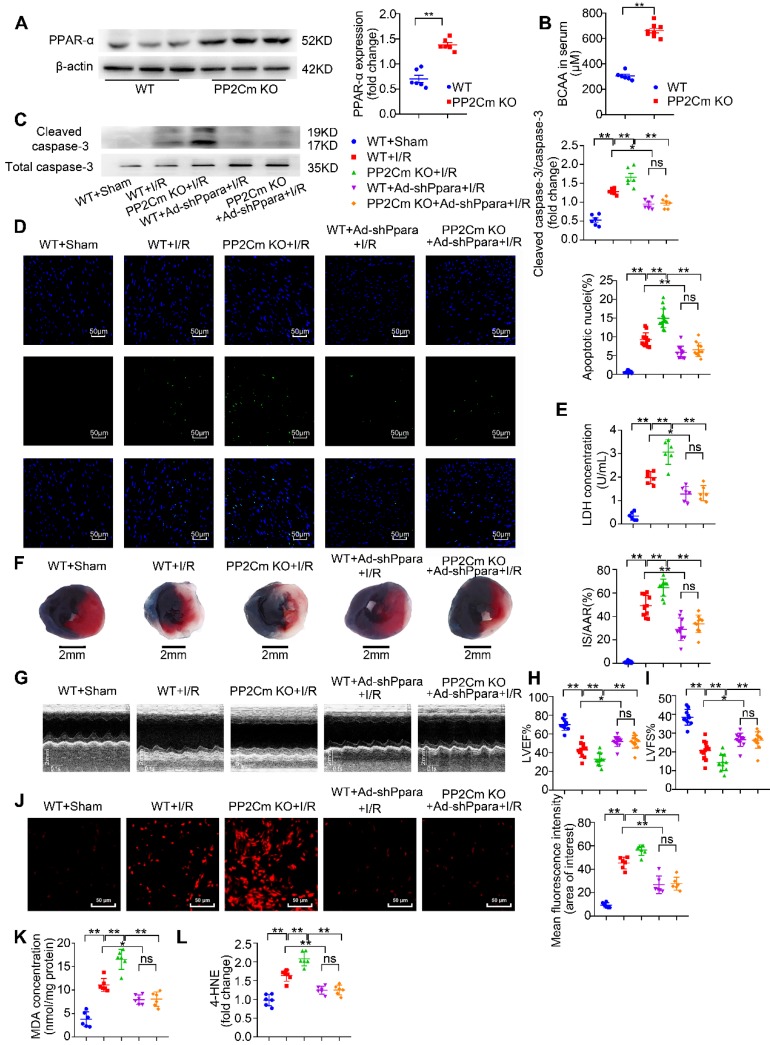
Rationale: Myocardial vulnerability to ischemia/reperfusion (I/R) injury is strictly regulated by energy substrate metabolism. Branched chain amino acids (BCAA), consisting of valine, leucine and isoleucine, are a group of essential amino acids that are highly oxidized in the heart. Elevated levels of BCAA have been implicated in the development of cardiovascular diseases; however, the role of BCAA in I/R process is not fully understood. The present study aims to determine how BCAA influence myocardial energy substrate metabolism and to further clarify the pathophysiological significance during cardiac I/R injury. Methods: Parameters of glucose and fatty acid metabolism were measured by seahorse metabolic flux analyzer in adult mouse cardiac myocytes with or without BCAA incubation. Chronic accumulation of BCAA was induced in mice receiving oral BCAA administration. A genetic mouse model with defective BCAA catabolism was also utilized. Mice were subjected to MI/R and the injury was assessed extensively at the whole-heart, cardiomyocyte, and molecular levels. Results: We confirmed that chronic accumulation of BCAA enhanced glycolysis and fatty acid oxidation (FAO) but suppressed glucose oxidation in adult mouse ventricular cardiomyocytes. Oral gavage of BCAA enhanced FAO in cardiac tissues, exacerbated lipid peroxidation toxicity and worsened myocardial vulnerability to I/R injury. Etomoxir, a specific inhibitor of FAO, rescued the deleterious effects of BCAA on I/R injury. Mechanistically, valine, leucine and their corresponding branched chain α-keto acid (BCKA) derivatives, but not isoleucine and its BCKA derivative, transcriptionally upregulated peroxisome proliferation-activated receptor alpha (PPAR-α). BCAA/BCKA induced PPAR-α upregulation through the general control nonderepresible-2 (GCN2)/ activating transcription factor-6 (ATF6) pathway. Finally, in a genetic mouse model with BCAA catabolic defects, chronic accumulation of BCAA increased FAO in myocardial tissues and sensitized the heart to I/R injury, which could be reversed by adenovirus-mediated PPAR-α silencing. Conclusions: We identify BCAA as an important nutrition regulator of myocardial fatty acid metabolism through transcriptional upregulation of PPAR-α. Chronic accumulation of BCAA, caused by either dietary or genetic factors, renders the heart vulnerable to I/R injury via exacerbating lipid peroxidation toxicity. These data support the notion that BCAA lowering methods might be potentially effective cardioprotective strategies, especially among patients with diseases characterized by elevated levels of BCAA, such as obesity and diabetes.
Keywords: Branched chain amino acids; Fatty acid metabolism; Ischemia/reperfusion injury; Peroxisome proliferation-activated receptor-α; Vulnerability..
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H. et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. - PubMed
-
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. - PubMed
-
- Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M. et al. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation. 2006;114:1721–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
