

EGFR-targeted CAR-T cells are potent and specific in suppressing triple-negative breast cancer both in vitro and in vivo
- PMID: 32373345
- PMCID: PMC7196685
- DOI: 10.1002/cti2.1135
EGFR-targeted CAR-T cells are potent and specific in suppressing triple-negative breast cancer both in vitro and in vivo
Abstract
Objectives: Triple-negative breast cancer (TNBC) is well known for its strong invasiveness, rapid recurrence and poor prognosis. Immunotherapy, including chimeric antigen receptor-modified T (CAR-T) cells, has emerged as a promising tool to treat TNBC. The identification of a specific target tumor antigen and the design of an effective CAR are among the many challenges of CAR-T therapy.
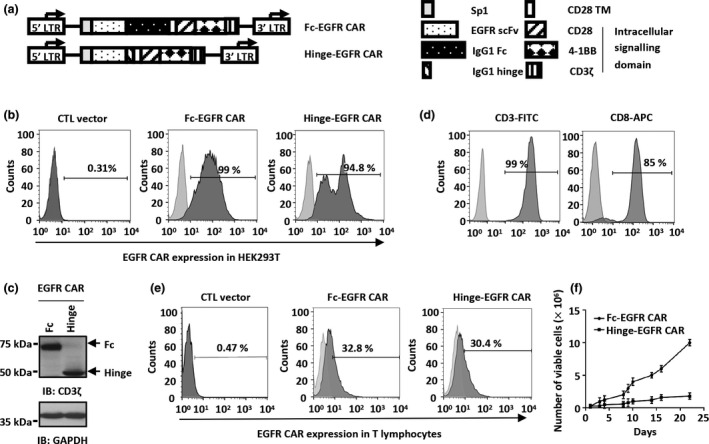
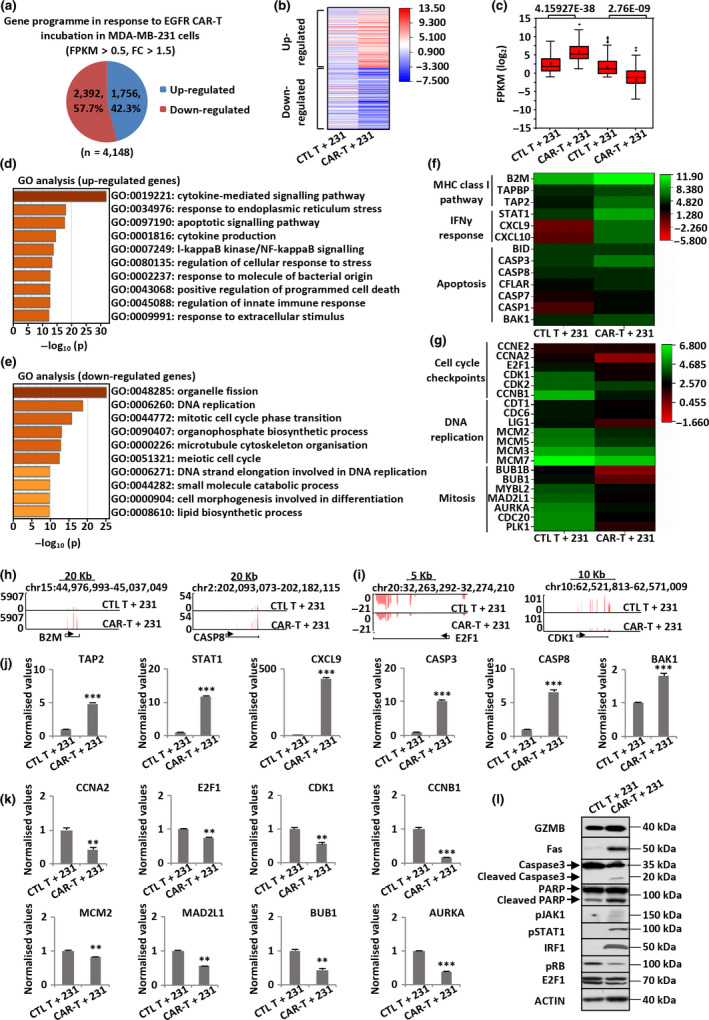
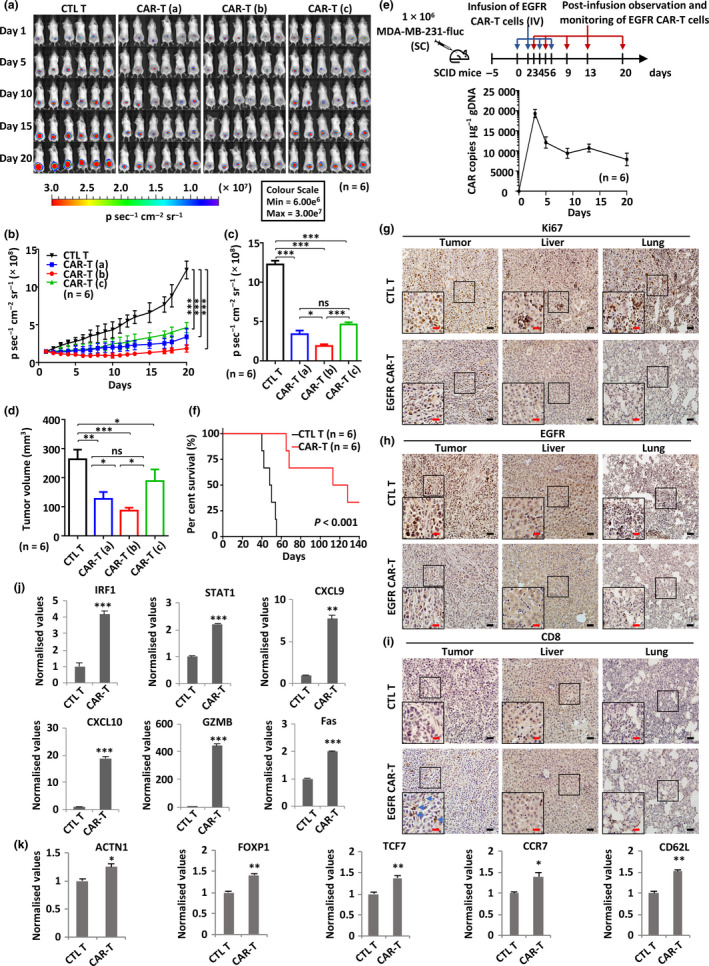
Methods: We reported that epidermal growth factor receptor (EGFR) is highly expressed in TNBC and consequently designed an optimal third generation of CAR targeting EGFR. The efficacy of primary T lymphocytes infected with EGFR CAR lentivirus (EGFR CAR-T) against TNBC was evaluated both in vitro and in vivo. The signalling pathways activated in tumor and EGFR CAR-T cells were revealed by RNA sequencing analysis.
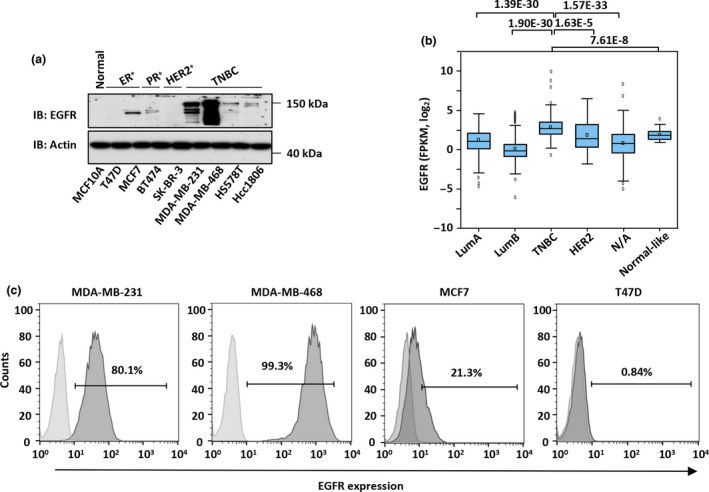
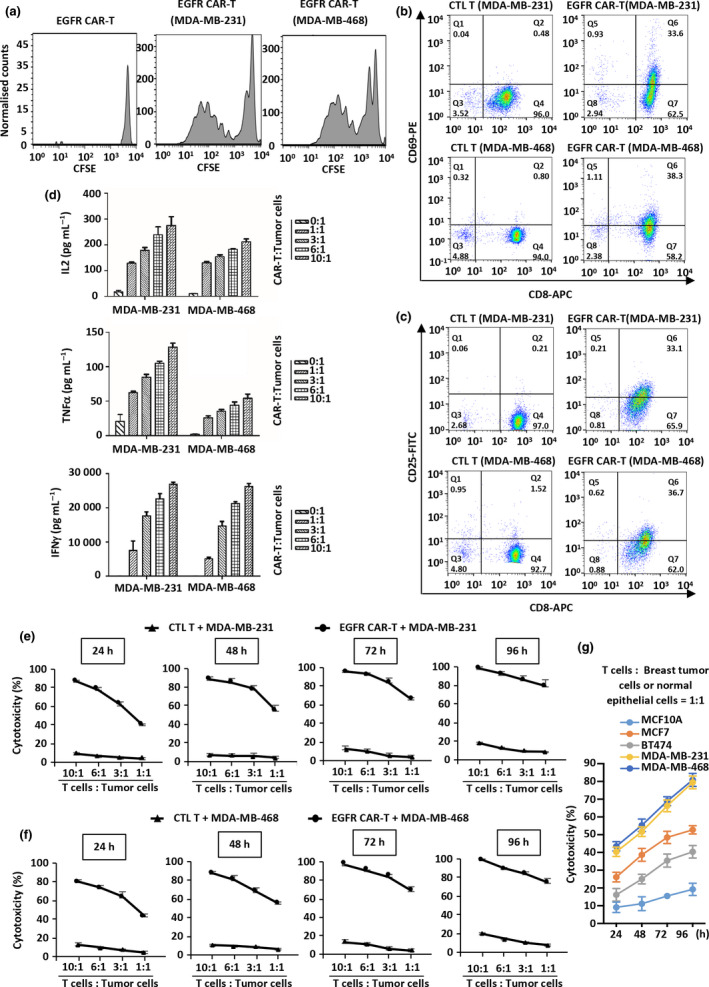
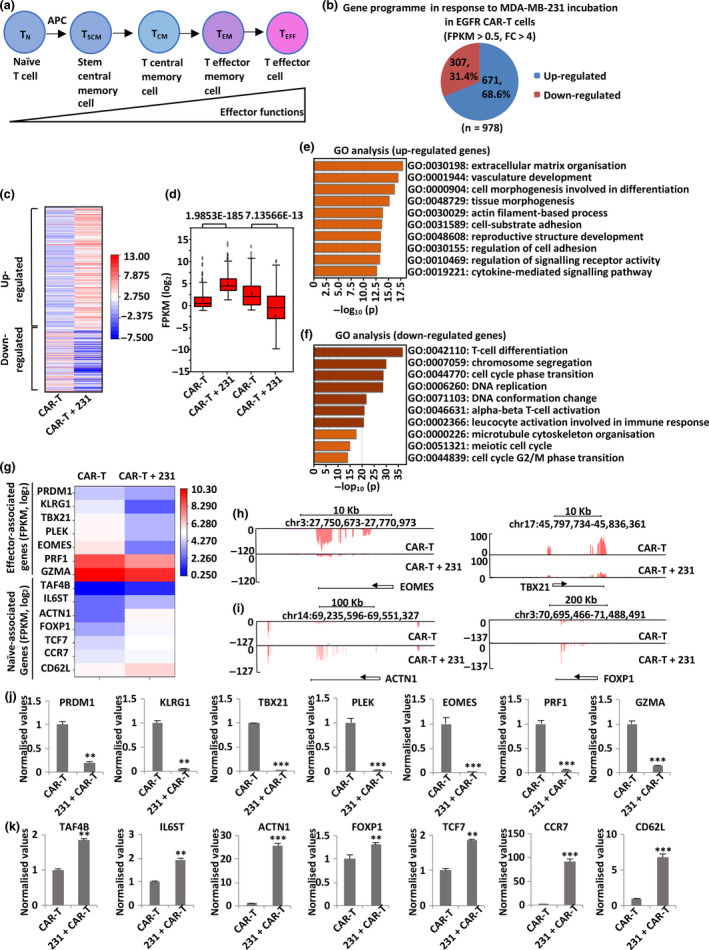
Results: Third-generation EGFR CAR-T cells exerted potent and specific suppression of TNBC cell growth in vitro, whereas limited cytotoxicity was observed towards normal breast epithelial cells or oestrogen receptor-positive breast cancer cells. This capability was further demonstrated in vivo in a xenograft mouse model, with minimal off-tumor cytotoxicity. Mechanistically, in vitro stimulation with TNBC cells induced the expansion of naïve-associated EGFR CAR-T cells and enhanced their persistence. Furthermore, EGFR CAR-T cells activated the interferon γ, granzyme-perforin-PARP and Fas-FADD-caspase signalling pathways in TNBC cells.
Conclusion: We demonstrate that EGFR is a relevant immunotherapeutic target in TNBC, and EGFR CAR-T exhibits potent and specific antitumor activity against TNBC, suggesting the potential of this third-generation EGFR CAR-T as an immunotherapy tool to treat TNBC in the clinic.
Keywords: chimeric antigen receptor‐modified T‐cell; epidermal growth factor receptor; immunotherapy; triple‐negative breast cancer.
© 2020 The Authors. Clinical & Translational Immunology published by John Wiley & Sons Australia, Ltd on behalf of Australian and New Zealand Society for Immunology, Inc.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Targeting epidermal growth factor-overexpressing triple-negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor.Cell Prolif. 2020 Aug;53(8):e12858. doi: 10.1111/cpr.12858. Epub 2020 Jun 27. Cell Prolif. 2020. PMID: 32592435 Free PMC article.
-
EGFR-specific CAR-T cells trigger cell lysis in EGFR-positive TNBC.Aging (Albany NY). 2019 Dec 4;11(23):11054-11072. doi: 10.18632/aging.102510. Epub 2019 Dec 4. Aging (Albany NY). 2019. PMID: 31804974 Free PMC article.
-
EGFR as a potent CAR T target in triple negative breast cancer brain metastases.Breast Cancer Res Treat. 2023 Jan;197(1):57-69. doi: 10.1007/s10549-022-06783-1. Epub 2022 Nov 1. Breast Cancer Res Treat. 2023. PMID: 36318382 Free PMC article.
-
Novel chimeric antigen receptor T cell-based immunotherapy: a perspective for triple-negative breast cancer.Front Cell Dev Biol. 2023 Jun 29;11:1158539. doi: 10.3389/fcell.2023.1158539. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37457288 Free PMC article. Review.
-
CAR-T cell therapy in triple-negative breast cancer: Hunting the invisible devil.Front Immunol. 2022 Nov 22;13:1018786. doi: 10.3389/fimmu.2022.1018786. eCollection 2022. Front Immunol. 2022. PMID: 36483567 Free PMC article. Review.
Cited by
-
Super-enhancer-controlled positive feedback loop BRD4/ERα-RET-ERα promotes ERα-positive breast cancer.Nucleic Acids Res. 2022 Oct 14;50(18):10230-10248. doi: 10.1093/nar/gkac778. Nucleic Acids Res. 2022. PMID: 36124682 Free PMC article.
-
Clinical significance and immune characteristics analysis of miR-221-3p and its key target genes related to epithelial-mesenchymal transition in breast cancer.Aging (Albany NY). 2024 Jan 6;16(1):322-347. doi: 10.18632/aging.205370. Epub 2024 Jan 6. Aging (Albany NY). 2024. PMID: 38189813 Free PMC article.
-
Immune gene therapy of cancer.Turk J Med Sci. 2020 Nov 3;50(SI-2):1679-1690. doi: 10.3906/sag-2005-327. Turk J Med Sci. 2020. PMID: 32512674 Free PMC article. Review.
-
Role of Gut Microbiota in Breast Cancer and Drug Resistance.Pathogens. 2023 Mar 16;12(3):468. doi: 10.3390/pathogens12030468. Pathogens. 2023. PMID: 36986390 Free PMC article. Review.
-
New progress in imaging diagnosis and immunotherapy of breast cancer.Front Immunol. 2025 Mar 17;16:1560257. doi: 10.3389/fimmu.2025.1560257. eCollection 2025. Front Immunol. 2025. PMID: 40165974 Free PMC article. Review.
References
-
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68: 394–424. - PubMed
-
- Perou CM, Sorlie T, Eisen MB et al Molecular portraits of human breast tumours. Nature 2000; 406: 747–752. - PubMed
-
- Foulkes WD, Smith Ian E, Reisfilho Jorge S. Triple‐negative breast cancer. N Engl J Med 2010; 363: 1938–1948. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous