

Animal Disease Surveillance in the 21st Century: Applications and Robustness of Phylodynamic Methods in Recent U.S. Human-Like H3 Swine Influenza Outbreaks
- PMID: 32373634
- PMCID: PMC7186338
- DOI: 10.3389/fvets.2020.00176
Animal Disease Surveillance in the 21st Century: Applications and Robustness of Phylodynamic Methods in Recent U.S. Human-Like H3 Swine Influenza Outbreaks
Abstract
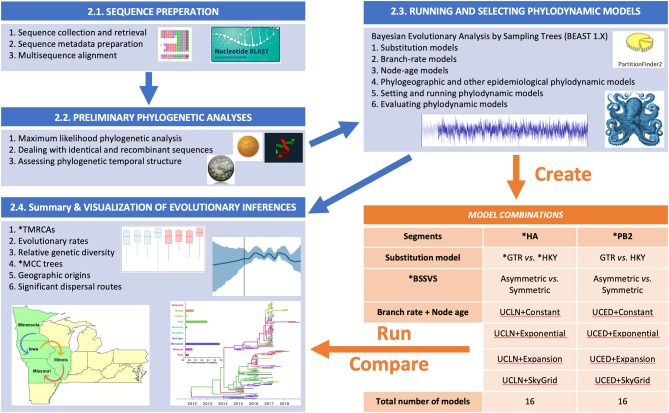
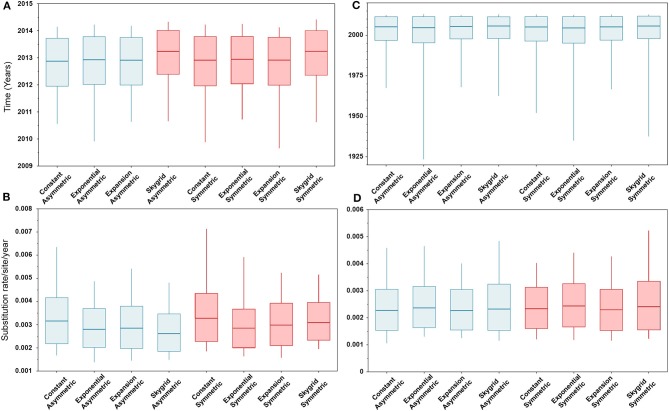
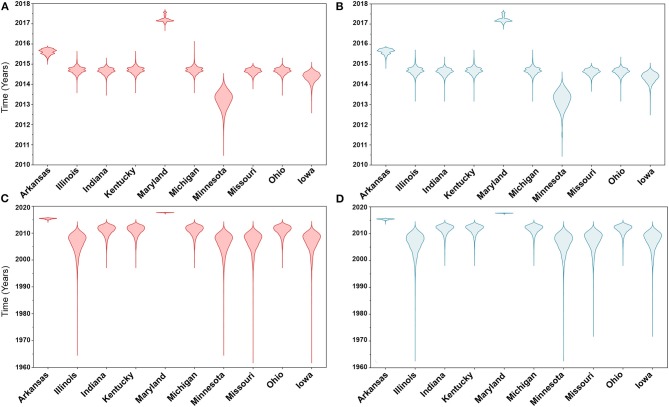
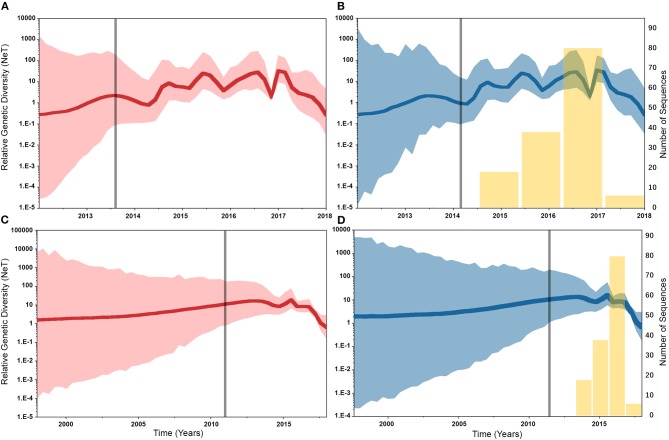
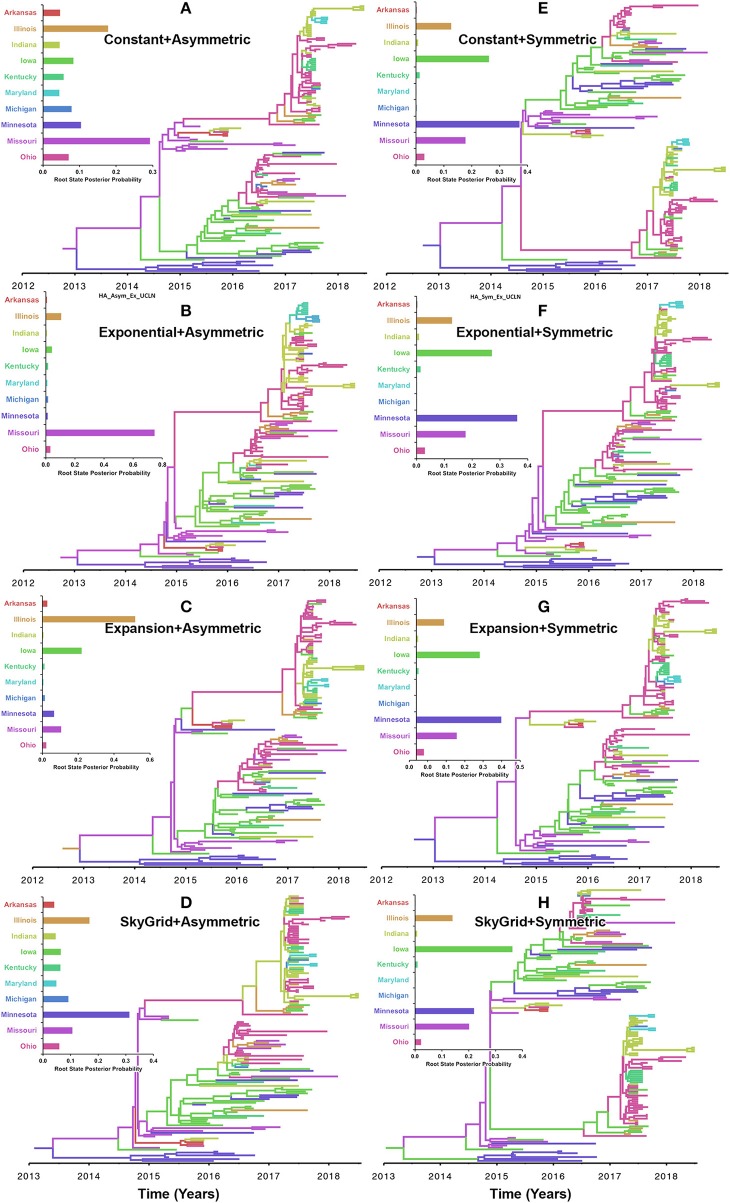
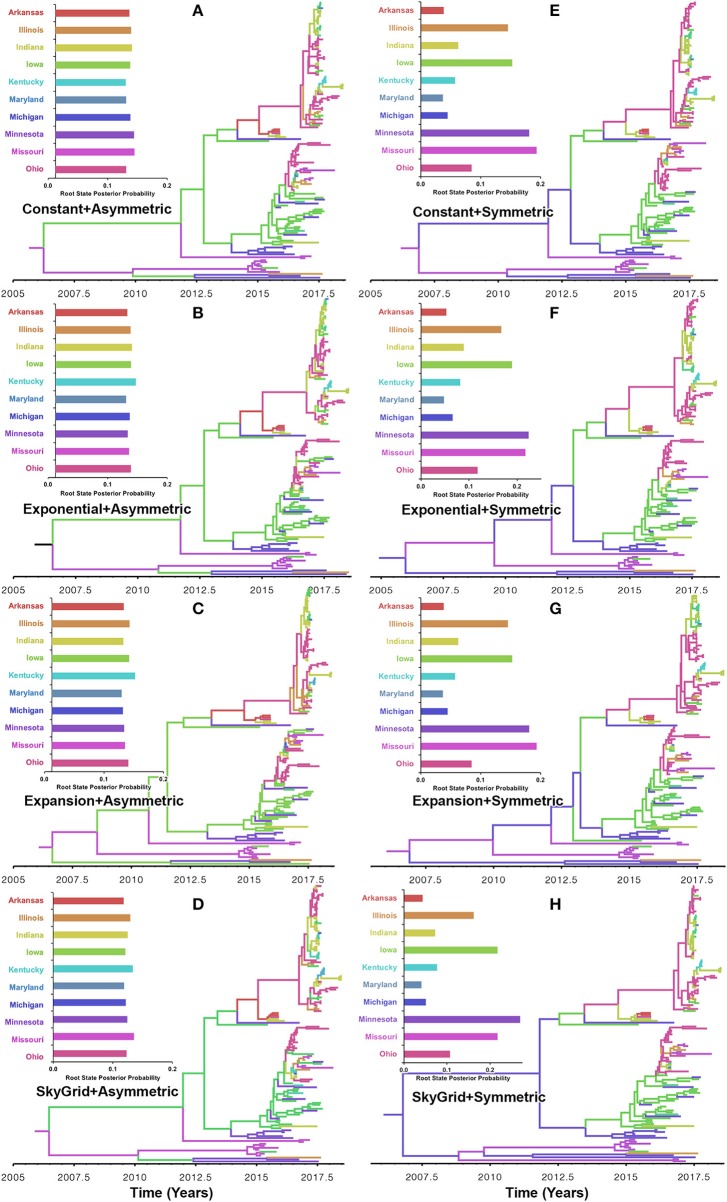
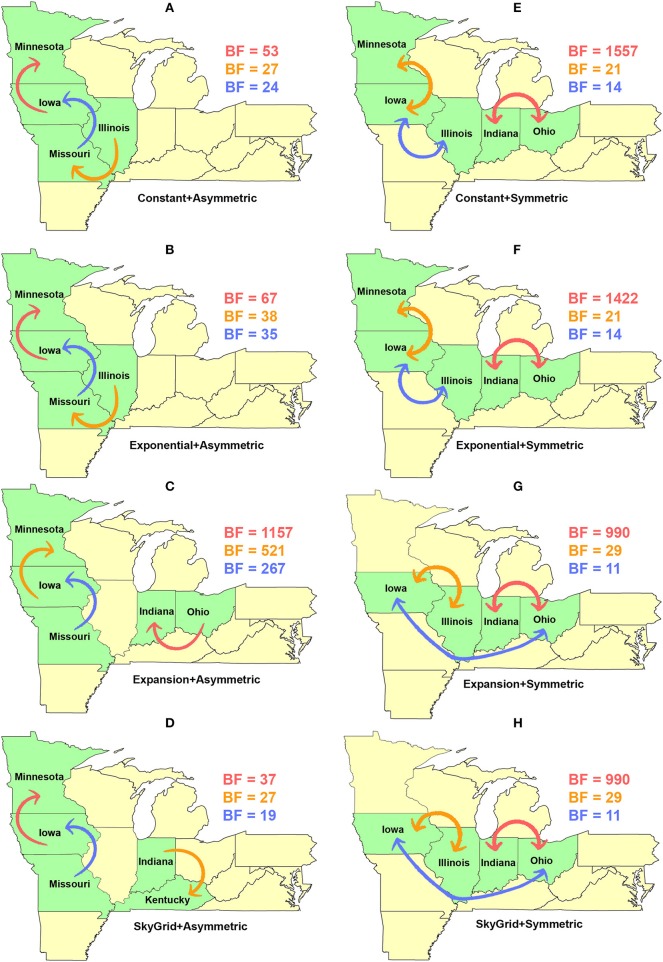
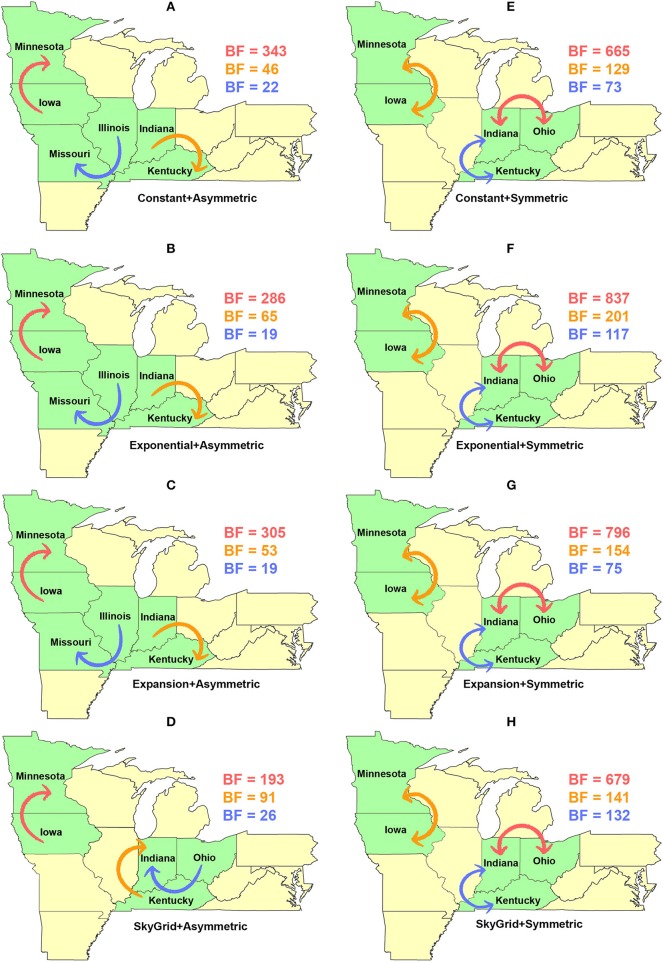
Emerging and endemic animal viral diseases continue to impose substantial impacts on animal and human health. Most current and past molecular surveillance studies of animal diseases investigated spatio-temporal and evolutionary dynamics of the viruses in a disjointed analytical framework, ignoring many uncertainties and made joint conclusions from both analytical approaches. Phylodynamic methods offer a uniquely integrated platform capable of inferring complex epidemiological and evolutionary processes from the phylogeny of viruses in populations using a single Bayesian statistical framework. In this study, we reviewed and outlined basic concepts and aspects of phylodynamic methods and attempted to summarize essential components of the methodology in one analytical pipeline to facilitate the proper use of the methods by animal health researchers. Also, we challenged the robustness of the posterior evolutionary parameters, inferred by the commonly used phylodynamic models, using hemagglutinin (HA) and polymerase basic 2 (PB2) segments of the currently circulating human-like H3 swine influenza (SI) viruses isolated in the United States and multiple priors. Subsequently, we compared similarities and differences between the posterior parameters inferred from sequence data using multiple phylodynamic models. Our suggested phylodynamic approach attempts to reduce the impact of its inherent limitations to offer less biased and biologically plausible inferences about the pathogen evolutionary characteristics to properly guide intervention activities. We also pinpointed requirements and challenges for integrating phylodynamic methods in routine animal disease surveillance activities.
Keywords: disease surveillance; evolutionary epidemiology; human-like H3; phylodynamics; phylogeography; swine influenza.
Copyright © 2020 Alkhamis, Li and Torremorell.
Figures
References
-
- Alvarez J, Valdes-Donoso P, Tousignant S, Alkhamis M, Morrison R, Perez A. Novel analytic tools for the study of porcine reproductive and respiratory syndrome virus (PRRSv) in endemic settings: lessons learned in the U.S. Porcine Health Manag. (2016) 2:1–9. 10.1186/s40813-016-0019-0 - DOI - PMC - PubMed
-
- Vilalta C, Arruda AG, Tousignant SJP, Valdes-Donoso P, Muellner P, Muellner U, et al. A Review of quantitative tools used to assess the epidemiology of porcine reproductive and respiratory syndrome in U.S. swine farms using dr. morrison's swine health monitoring program data. Front Vet Sci. (2017) 4:94. 10.3389/fvets.2017.00094 - DOI - PMC - PubMed
Associated data
LinkOut - more resources
Full Text Sources
Miscellaneous