

Differential activity and selectivity of N-terminal modified CXCL12 chemokines at the CXCR4 and ACKR3 receptors
- PMID: 32374043
- PMCID: PMC7540625
- DOI: 10.1002/JLB.2MA0320-383RR
Differential activity and selectivity of N-terminal modified CXCL12 chemokines at the CXCR4 and ACKR3 receptors
Abstract
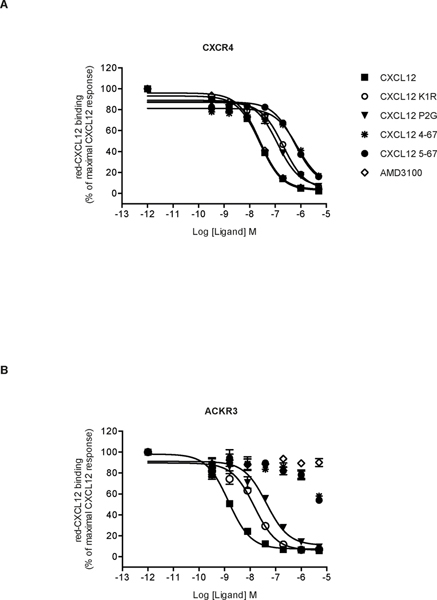
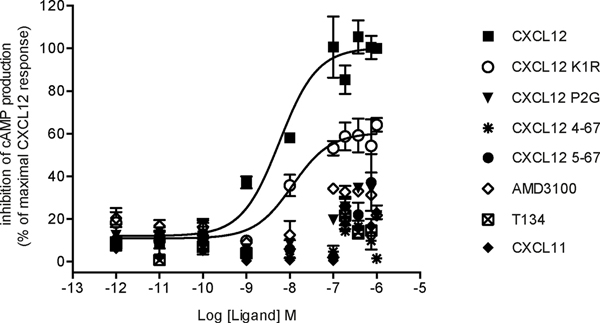
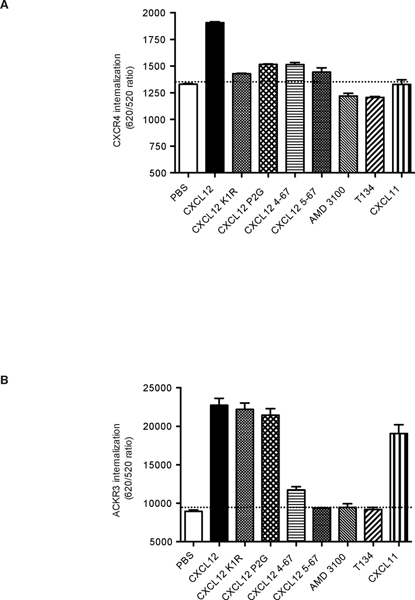
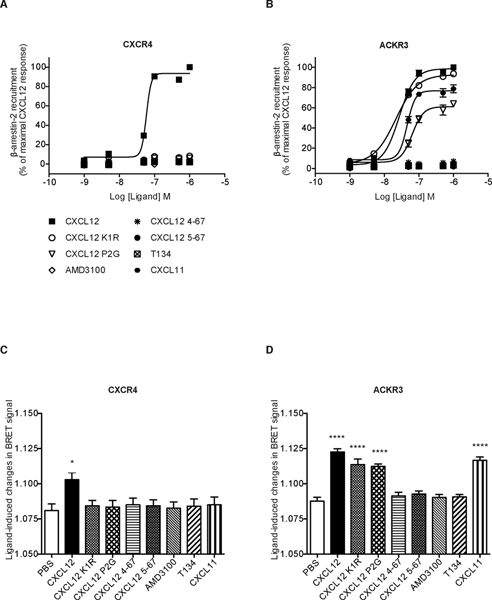
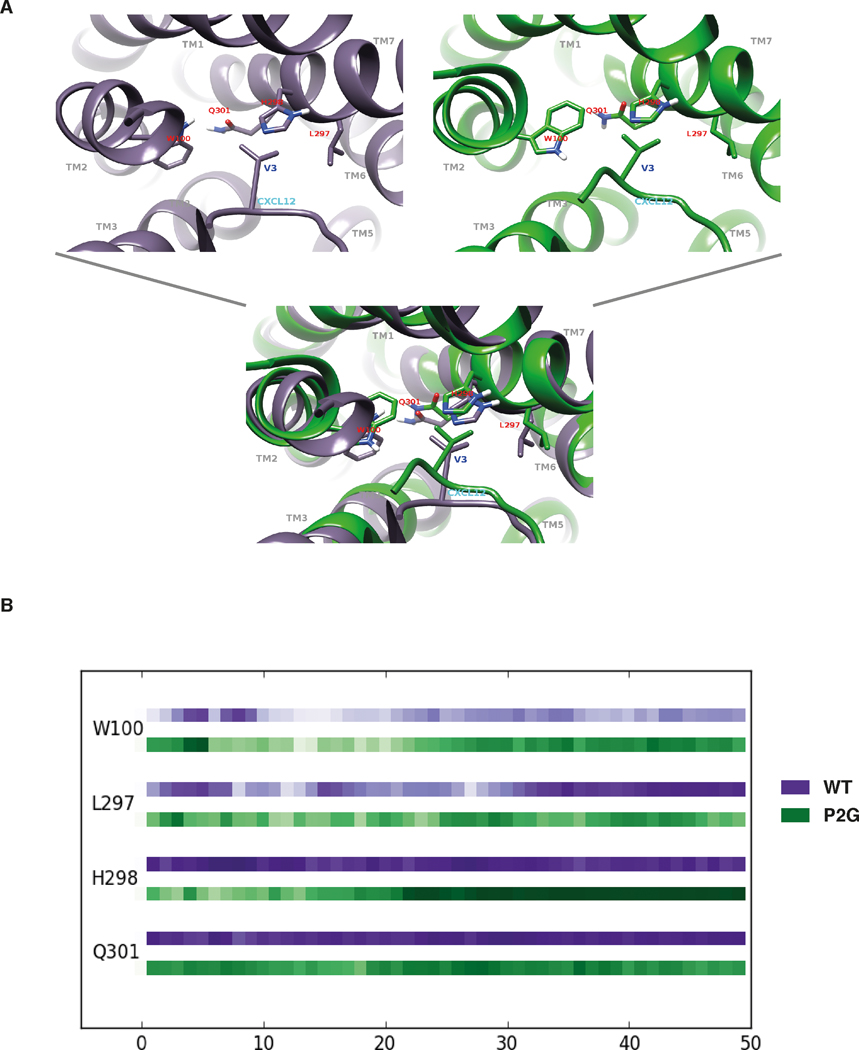
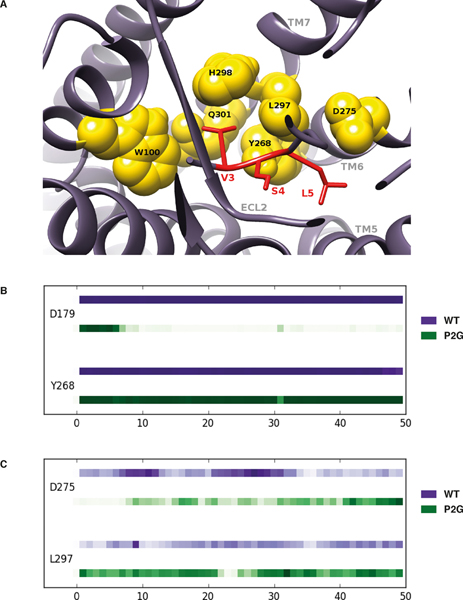
Chemokines play critical roles in numerous physiologic and pathologic processes through their action on seven-transmembrane (TM) receptors. The N-terminal domain of chemokines, which is a key determinant of signaling via its binding within a pocket formed by receptors' TM helices, can be the target of proteolytic processing. An illustrative case of this regulatory mechanism is the natural processing of CXCL12 that generates chemokine variants lacking the first two N-terminal residues. Whereas such truncated variants behave as antagonists of CXCR4, the canonical G protein-coupled receptor of CXCL12, they are agonists of the atypical chemokine receptor 3 (ACKR3/CXCR7), suggesting the implication of different structural determinants in the complexes formed between CXCL12 and its two receptors. Recent analyses have suggested that the CXCL12 N-terminus first engages the TM helices of ACKR3 followed by the receptor N-terminus wrapping around the chemokine core. Here we investigated the first stage of ACKR3-CXCL12 interactions by comparing the activity of substituted or N-terminally truncated variants of CXCL12 toward CXCR4 and ACKR3. We showed that modification of the first two N-terminal residues of the chemokine (K1R or P2G) does not alter the ability of CXCL12 to activate ACKR3. Our results also identified the K1R variant as a G protein-biased agonist of CXCR4. Comparative molecular dynamics simulations of the complexes formed by ACKR3 either with CXCL12 or with the P2G variant identified interactions between the N-terminal 2-4 residues of CXCL12 and a pocket formed by receptor's TM helices 2, 6, and 7 as critical determinants for ACKR3 activation.
Keywords: ACKR3; CXCL12; CXCR4; GPCR signaling; chemokine variants; pluridimensional efficacy.
©2020 Society for Leukocyte Biology.
Conflict of interest statement
DISCLOSURE
The authors declare that they have no conflict of interest.
Figures

Similar articles
-
Kinetics of CXCL12 binding to atypical chemokine receptor 3 reveal a role for the receptor N terminus in chemokine binding.Sci Signal. 2019 Sep 10;12(598):eaaw3657. doi: 10.1126/scisignal.aaw3657. Sci Signal. 2019. PMID: 31506383 Free PMC article.
-
Dual targeting of the chemokine receptors CXCR4 and ACKR3 with novel engineered chemokines.J Biol Chem. 2015 Sep 11;290(37):22385-97. doi: 10.1074/jbc.M115.675108. Epub 2015 Jul 27. J Biol Chem. 2015. PMID: 26216880 Free PMC article.
-
Mutational Analysis of Atypical Chemokine Receptor 3 (ACKR3/CXCR7) Interaction with Its Chemokine Ligands CXCL11 and CXCL12.J Biol Chem. 2017 Jan 6;292(1):31-42. doi: 10.1074/jbc.M116.762252. Epub 2016 Nov 14. J Biol Chem. 2017. PMID: 27875312 Free PMC article.
-
The relevance of the chemokine receptor ACKR3/CXCR7 on CXCL12-mediated effects in cancers with a focus on virus-related cancers.Cytokine Growth Factor Rev. 2014 Jun;25(3):307-16. doi: 10.1016/j.cytogfr.2014.04.006. Epub 2014 May 9. Cytokine Growth Factor Rev. 2014. PMID: 24853339 Review.
-
CXCL12-CXCR4/CXCR7 Axis in Colorectal Cancer: Therapeutic Target in Preclinical and Clinical Studies.Int J Mol Sci. 2021 Jul 9;22(14):7371. doi: 10.3390/ijms22147371. Int J Mol Sci. 2021. PMID: 34298991 Free PMC article. Review.
Cited by
-
Discovery of Bis-Imidazoline Derivatives as New CXCR4 Ligands.Molecules. 2023 Jan 24;28(3):1156. doi: 10.3390/molecules28031156. Molecules. 2023. PMID: 36770826 Free PMC article.
-
CXC Chemokine Ligand 12 Facilitates Gi Protein Binding to CXC Chemokine Receptor 4 by Stabilizing Packing of the Proline-Isoleucine-Phenylalanine Motif: Insights from Automated Path Searching.J Am Chem Soc. 2025 Mar 26;147(12):10129-10138. doi: 10.1021/jacs.4c14293. Epub 2025 Mar 17. J Am Chem Soc. 2025. PMID: 40096846 Free PMC article.
-
Immunoexpression of CXCL12 and CXCR4 in sporadic and Gorlin-Goltz syndrome-related odontogenic keratocysts.J Clin Exp Dent. 2022 May 1;14(5):e426-e432. doi: 10.4317/jced.59561. eCollection 2022 May. J Clin Exp Dent. 2022. PMID: 35582358 Free PMC article.
-
Distinct activation mechanisms of CXCR4 and ACKR3 revealed by single-molecule analysis of their conformational landscapes.Elife. 2025 Apr 15;13:RP100098. doi: 10.7554/eLife.100098. Elife. 2025. PMID: 40232828 Free PMC article.
-
Noncanonical roles of chemokine regions in CCR9 activation revealed by structural modeling and mutational mapping.Nat Commun. 2025 Aug 18;16(1):7695. doi: 10.1038/s41467-025-62321-9. Nat Commun. 2025. PMID: 40825773 Free PMC article.
References
-
- Janssens R, Struyf S, Proost P. (2018) Pathological roles of the homeostatic chemokine CXCL12. Cytokine & growth factor reviews 44, 51–68. - PubMed
-
- Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, Schall TJ (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med 203, 2201–13. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
