

The genomic landscape of metastatic breast cancer: Insights from 11,000 tumors
- PMID: 32374727
- PMCID: PMC7202592
- DOI: 10.1371/journal.pone.0231999
The genomic landscape of metastatic breast cancer: Insights from 11,000 tumors
Abstract
Background: Metastatic breast cancer is the leading cause of cancer death in women, but the genomics of metastasis in breast cancer are poorly studied.
Methods: We explored a set of 11,616 breast tumors, including 5,034 metastases, which had undergone targeted sequencing during standard clinical care.
Results: Besides the known hotspot mutations in ESR1, we observed a metastatic enrichment of previously unreported, lower-prevalence mutations in the ligand-binding domain, implying that these mutations may also be functional. Furthermore, individual ESR1 hotspots are significantly enriched in specific metastatic tissues and histologies, suggesting functional differences between these mutations. Other alterations enriched across all metastases include loss of function of the CDK4 regulator CDKN1B, and mutations in the transcription factor CTCF. Mutations enriched at specific metastatic sites generally reflect biology of the target tissue and may be adaptations to growth in the local environment. These include PTEN and ASXL1 alterations in brain metastases and NOTCH1 alterations in skin. We observed an enrichment of KRAS, KEAP1, STK11 and EGFR mutations in lung metastases. However, the patterns of other mutations in these tumors indicate that these are misdiagnosed lung primaries rather than breast metastases.
Conclusions: An order-of-magnitude increase in samples relative to previous studies allowed us to detect novel genomic characteristics of metastatic cancer and to expand and clarify previous findings.
Conflict of interest statement
All authors were employees and may have held shares of Genentech/Roche or Foundation Medicine at the time this work was completed. Both companies are involved in the development of cancer drugs and clinical assays for metastatic breast cancer, and have multiple patents in related areas. This does not alter our adherence to PLOS ONE policies on sharing data and materials.
Figures
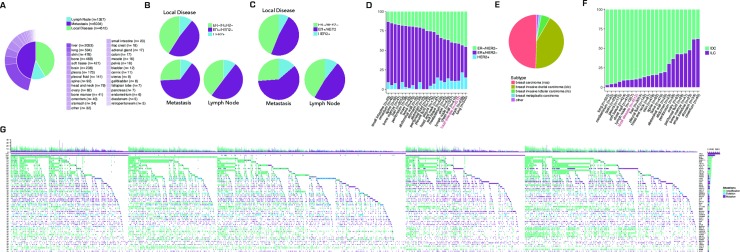
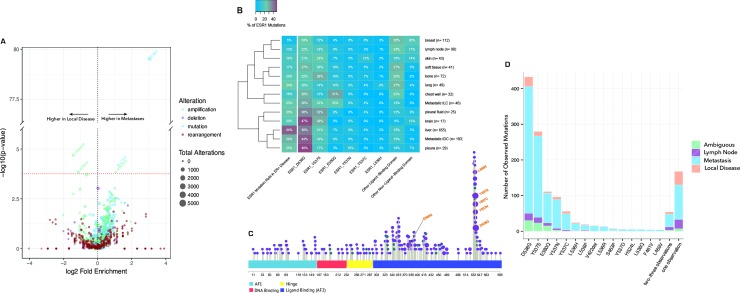
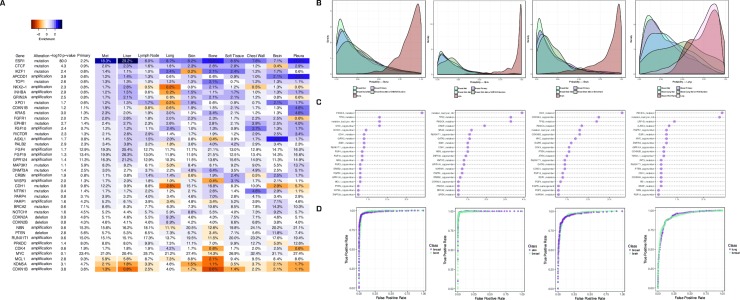
References
-
- Torre L. A., Islami F., Siegel R. L., Ward E. M. & Jemal A. Global cancer in women: burden and trends. (AACR, 2017). - PubMed
-
- Scully O. J., Bay B.-H., Yip G. & Yu Y. Breast cancer metastasis. Cancer Genomics-Proteomics 9, 311–320 (2012). - PubMed
-
- Jatoi I. & Rody A. Management of Breast Diseases. (Springer, 2016).
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
