

Multifaceted analysis of training and testing convolutional neural networks for protein secondary structure prediction
- PMID: 32374785
- PMCID: PMC7202669
- DOI: 10.1371/journal.pone.0232528
Multifaceted analysis of training and testing convolutional neural networks for protein secondary structure prediction
Abstract
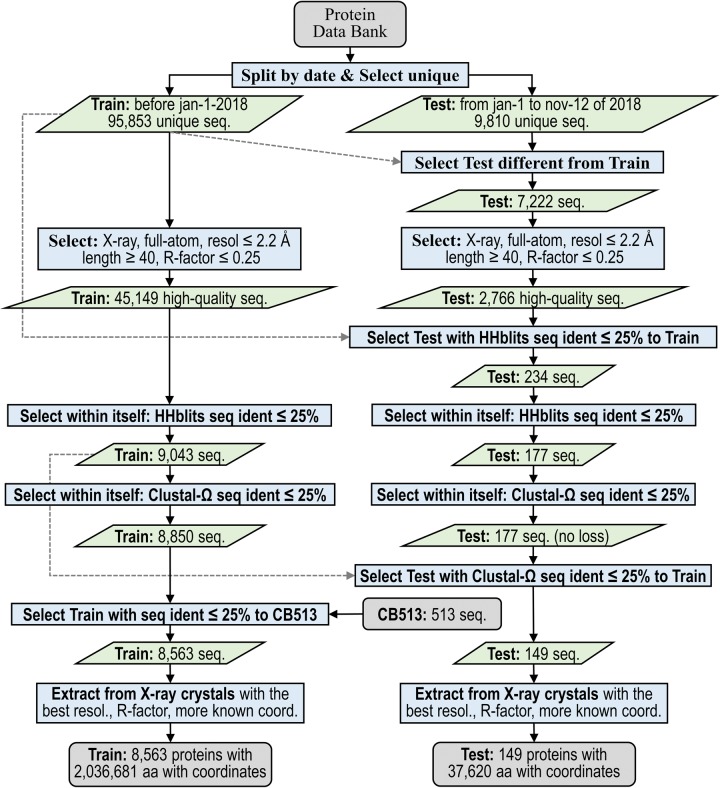
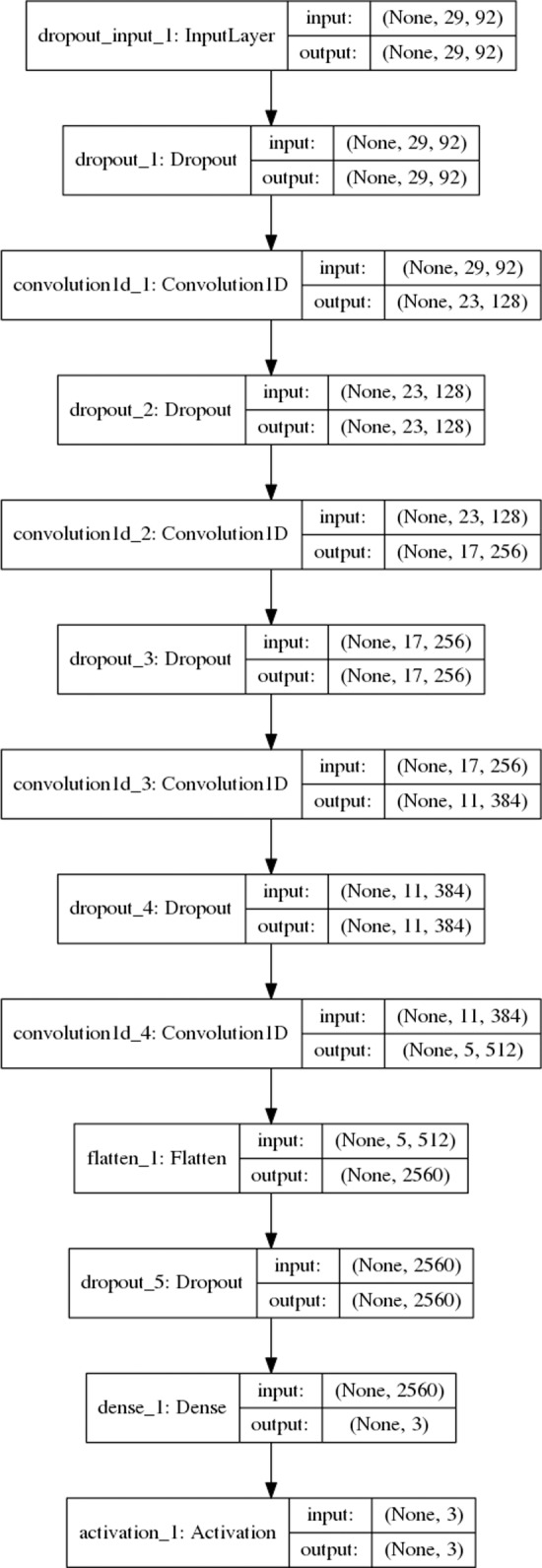
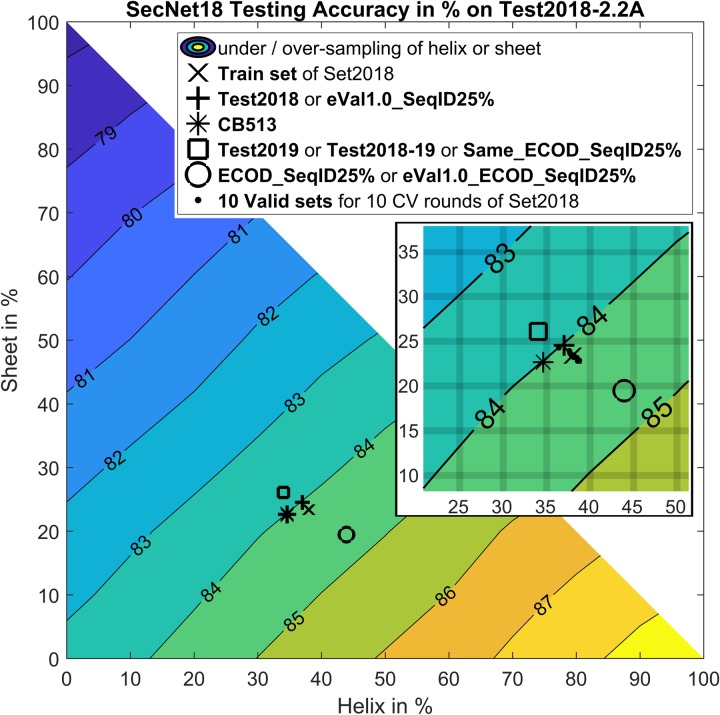
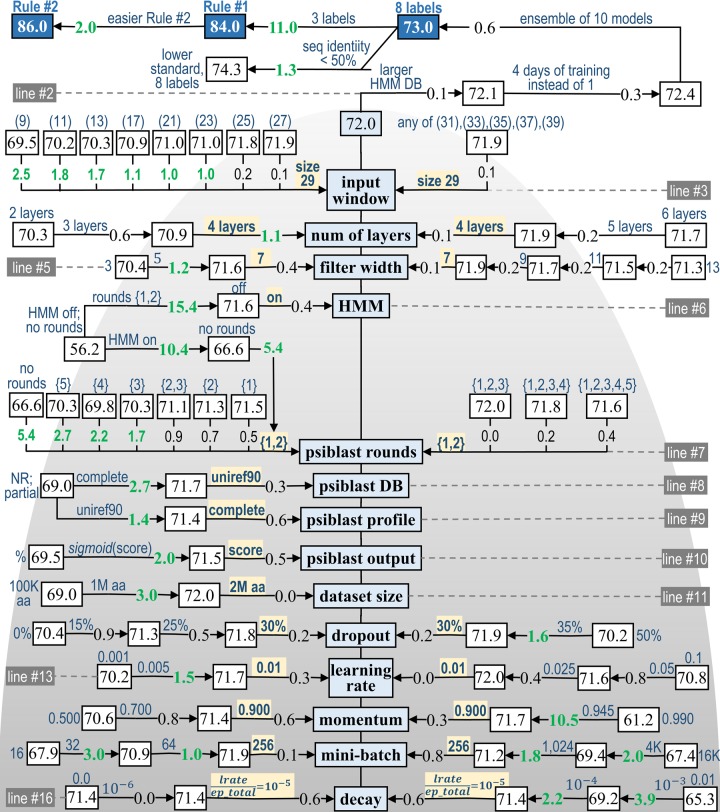
Protein secondary structure prediction remains a vital topic with broad applications. Due to lack of a widely accepted standard in secondary structure predictor evaluation, a fair comparison of predictors is challenging. A detailed examination of factors that contribute to higher accuracy is also lacking. In this paper, we present: (1) new test sets, Test2018, Test2019, and Test2018-2019, consisting of proteins from structures released in 2018 and 2019 with less than 25% identity to any protein published before 2018; (2) a 4-layer convolutional neural network, SecNet, with an input window of ±14 amino acids which was trained on proteins ≤25% identical to proteins in Test2018 and the commonly used CB513 test set; (3) an additional test set that shares no homologous domains with the training set proteins, according to the Evolutionary Classification of Proteins (ECOD) database; (4) a detailed ablation study where we reverse one algorithmic choice at a time in SecNet and evaluate the effect on the prediction accuracy; (5) new 4- and 5-label prediction alphabets that may be more practical for tertiary structure prediction methods. The 3-label accuracy (helix, sheet, coil) of the leading predictors on both Test2018 and CB513 is 81-82%, while SecNet's accuracy is 84% for both sets. Accuracy on the non-homologous ECOD set is only 0.6 points (83.9%) lower than the results on the Test2018-2019 set (84.5%). The ablation study of features, neural network architecture, and training hyper-parameters suggests the best accuracy results are achieved with good choices for each of them while the neural network architecture is not as critical as long as it is not too simple. Protocols for generating and using unbiased test, validation, and training sets are provided. Our data sets, including input features and assigned labels, and SecNet software including third-party dependencies and databases, are downloadable from dunbrack.fccc.edu/ss and github.com/sh-maxim/ss.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
