

Lysosomal Biology and Function: Modern View of Cellular Debris Bin
- PMID: 32375321
- PMCID: PMC7290337
- DOI: 10.3390/cells9051131
Lysosomal Biology and Function: Modern View of Cellular Debris Bin
Abstract
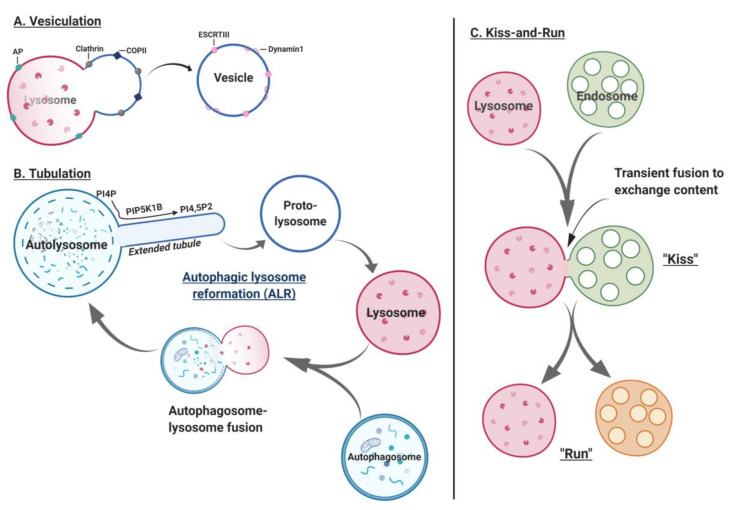
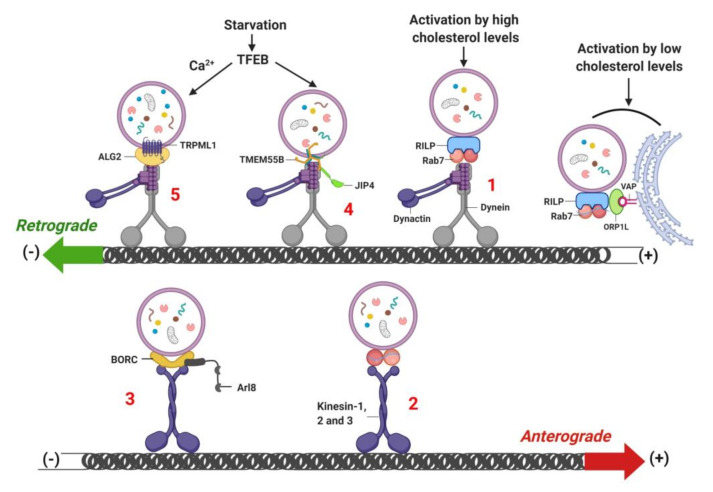
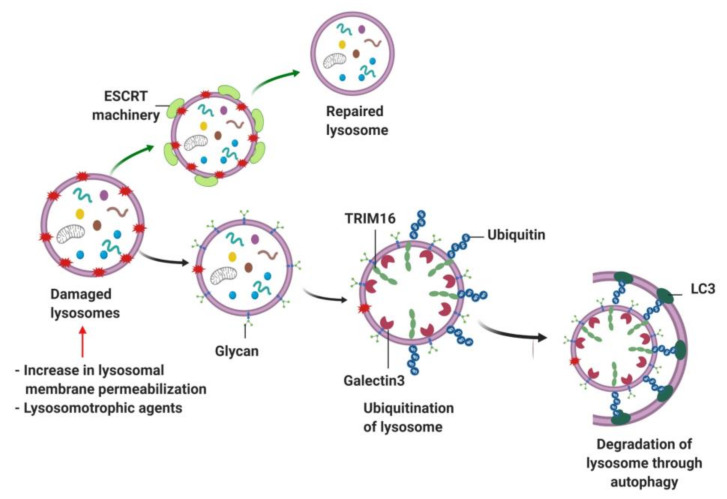
Lysosomes are the main proteolytic compartments of mammalian cells comprising of a battery of hydrolases. Lysosomes dispose and recycle extracellular or intracellular macromolecules by fusing with endosomes or autophagosomes through specific waste clearance processes such as chaperone-mediated autophagy or microautophagy. The proteolytic end product is transported out of lysosomes via transporters or vesicular membrane trafficking. Recent studies have demonstrated lysosomes as a signaling node which sense, adapt and respond to changes in substrate metabolism to maintain cellular function. Lysosomal dysfunction not only influence pathways mediating membrane trafficking that culminate in the lysosome but also govern metabolic and signaling processes regulating protein sorting and targeting. In this review, we describe the current knowledge of lysosome in influencing sorting and nutrient signaling. We further present a mechanistic overview of intra-lysosomal processes, along with extra-lysosomal processes, governing lysosomal fusion and fission, exocytosis, positioning and membrane contact site formation. This review compiles existing knowledge in the field of lysosomal biology by describing various lysosomal events necessary to maintain cellular homeostasis facilitating development of therapies maintaining lysosomal function.
Keywords: autophagy; calcium; cathepsin; endocytosis; lysosome; mannose-6-phosphate; metabolism; proton.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures
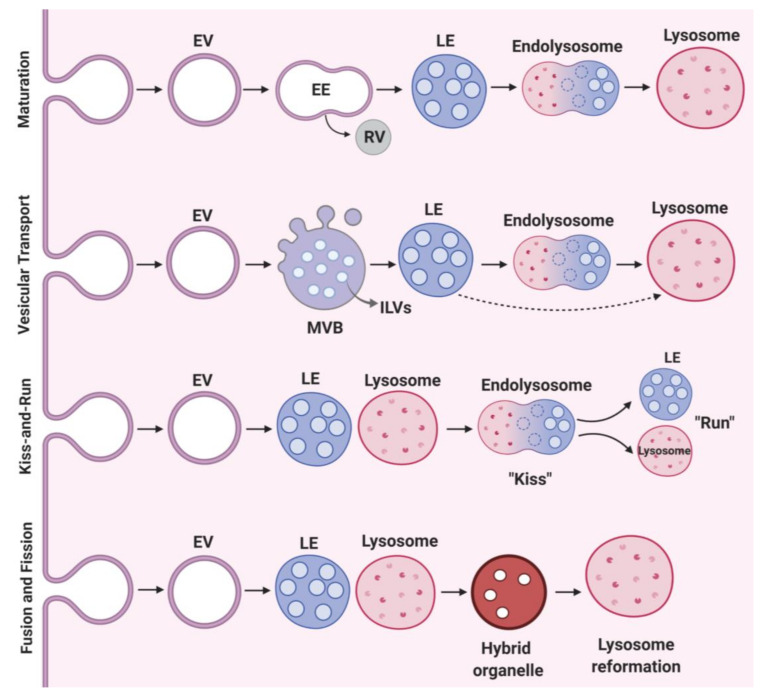
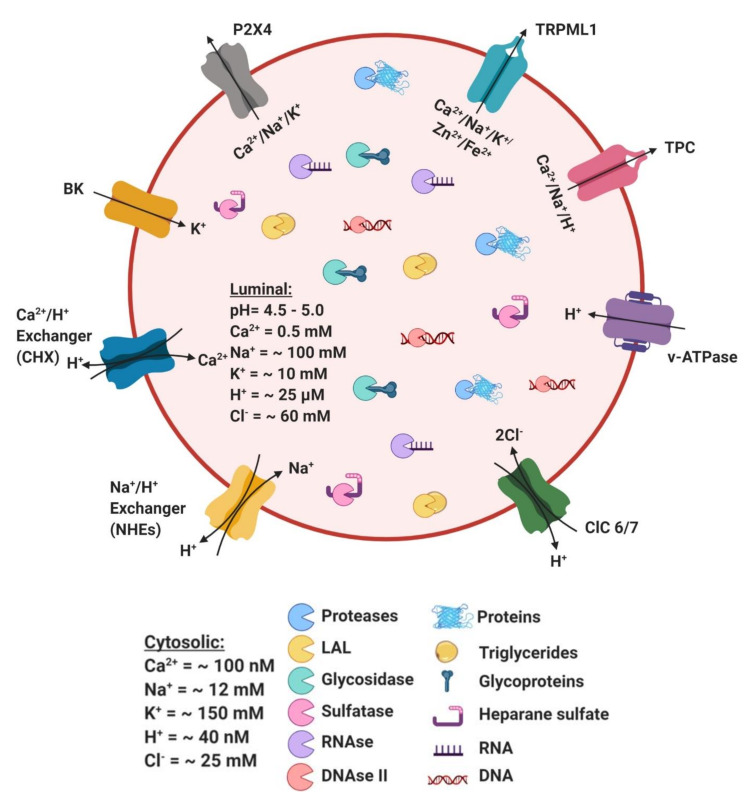
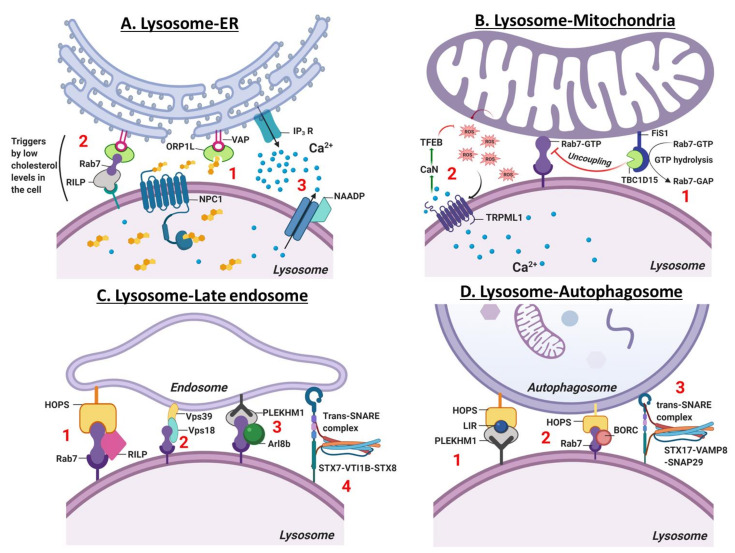
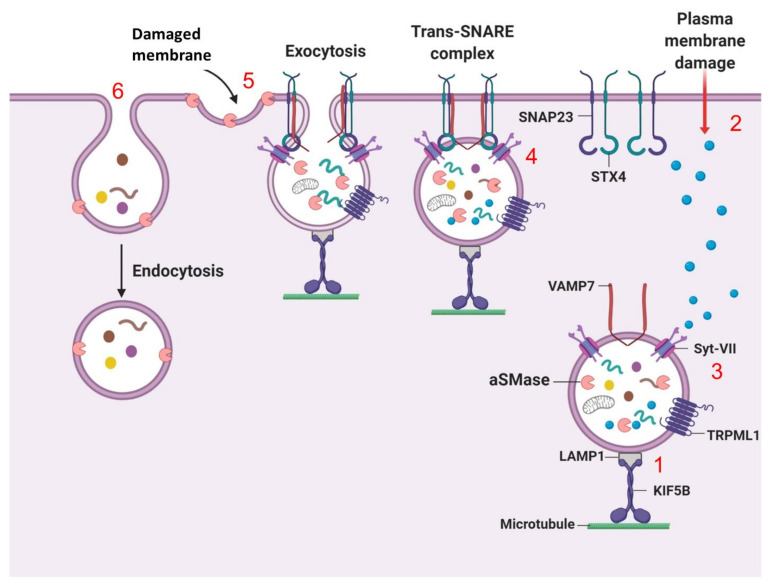
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
