

An Extensive Meta-Metagenomic Search Identifies SARS-CoV-2-Homologous Sequences in Pangolin Lung Viromes
- PMID: 32376697
- PMCID: PMC7203451
- DOI: 10.1128/mSphere.00160-20
An Extensive Meta-Metagenomic Search Identifies SARS-CoV-2-Homologous Sequences in Pangolin Lung Viromes
Abstract
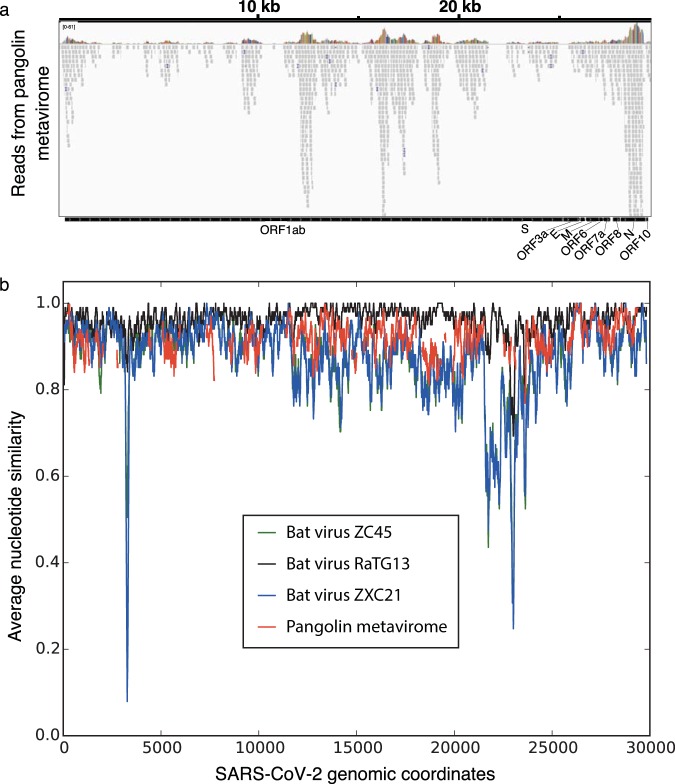
In numerous instances, tracking the biological significance of a nucleic acid sequence can be augmented through the identification of environmental niches in which the sequence of interest is present. Many metagenomic data sets are now available, with deep sequencing of samples from diverse biological niches. While any individual metagenomic data set can be readily queried using web-based tools, meta-searches through all such data sets are less accessible. In this brief communication, we demonstrate such a meta-metagenomic approach, examining close matches to the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in all high-throughput sequencing data sets in the NCBI Sequence Read Archive accessible with the "virome" keyword. In addition to the homology to bat coronaviruses observed in descriptions of the SARS-CoV-2 sequence (F. Wu, S. Zhao, B. Yu, Y. M. Chen, et al., Nature 579:265-269, 2020, https://doi.org/10.1038/s41586-020-2008-3; P. Zhou, X. L. Yang, X. G. Wang, B. Hu, et al., Nature 579:270-273, 2020, https://doi.org/10.1038/s41586-020-2012-7), we note a strong homology to numerous sequence reads in metavirome data sets generated from the lungs of deceased pangolins reported by Liu et al. (P. Liu, W. Chen, and J. P. Chen, Viruses 11:979, 2019, https://doi.org/10.3390/v11110979). While analysis of these reads indicates the presence of a similar viral sequence in pangolin lung, the similarity is not sufficient to either confirm or rule out a role for pangolins as an intermediate host in the recent emergence of SARS-CoV-2. In addition to the implications for SARS-CoV-2 emergence, this study illustrates the utility and limitations of meta-metagenomic search tools in effective and rapid characterization of potentially significant nucleic acid sequences.IMPORTANCE Meta-metagenomic searches allow for high-speed, low-cost identification of potentially significant biological niches for sequences of interest.
Keywords: COVID; SARS-nCoV-2; bioinformatics; coronavirus; metagenomics; pangolin.
Copyright © 2020 Wahba et al.
Figures
References
-
- Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. doi: 10.1038/s41586-020-2202-3. - DOI - PMC - PubMed
-
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi: 10.1038/s41586-020-2012-7. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
