

The role of mitochondrial reactive oxygen species, NO and H2 S in ischaemia/reperfusion injury and cardioprotection
- PMID: 32383522
- PMCID: PMC7299678
- DOI: 10.1111/jcmm.15279
The role of mitochondrial reactive oxygen species, NO and H2 S in ischaemia/reperfusion injury and cardioprotection
Abstract
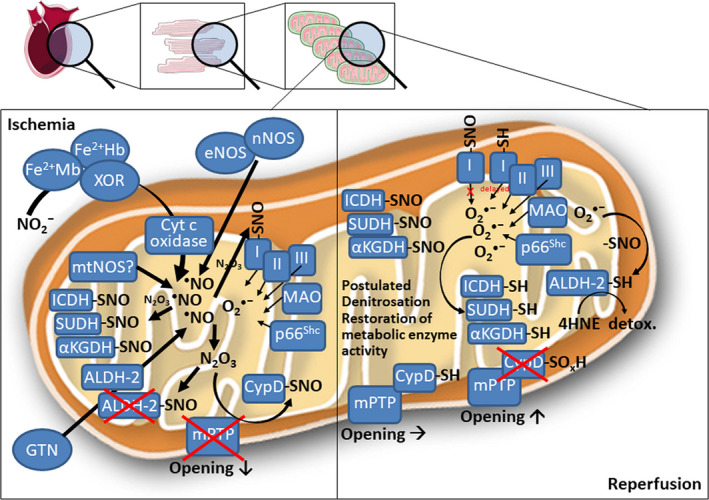
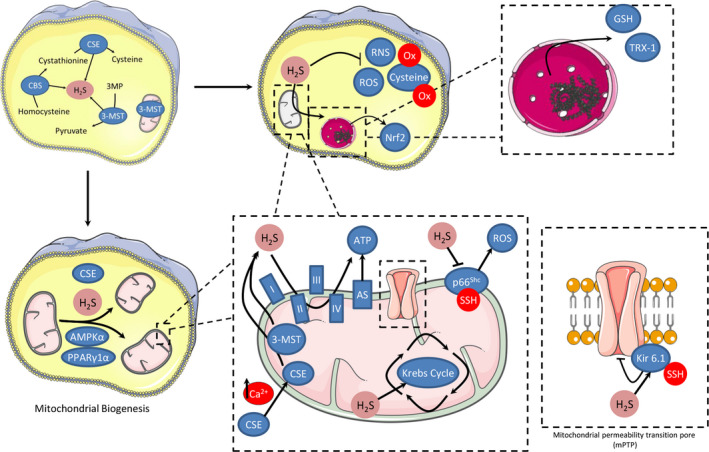
Redox signalling in mitochondria plays an important role in myocardial ischaemia/reperfusion (I/R) injury and in cardioprotection. Reactive oxygen and nitrogen species (ROS/RNS) modify cellular structures and functions by means of covalent changes in proteins including among others S-nitros(yl)ation by nitric oxide (NO) and its derivatives, and S-sulphydration by hydrogen sulphide (H2 S). Many enzymes are involved in the mitochondrial formation and handling of ROS, NO and H2 S under physiological and pathological conditions. In particular, the balance between formation and removal of reactive species is impaired during I/R favouring their accumulation. Therefore, various interventions aimed at decreasing mitochondrial ROS accumulation have been developed and have shown cardioprotective effects in experimental settings. However, ROS, NO and H2 S play also a role in endogenous cardioprotection, as in the case of ischaemic pre-conditioning, so that preventing their increase might hamper self-defence mechanisms. The aim of the present review was to provide a critical analysis of formation and role of reactive species, NO and H2 S in mitochondria, with a special emphasis on mechanisms of injury and protection that determine the fate of hearts subjected to I/R. The elucidation of the signalling pathways of ROS, NO and H2 S is likely to reveal novel molecular targets for cardioprotection that could be modulated by pharmacological agents to prevent I/R injury.
Keywords: cardioprotection; heart; hydrogen sulphide; ischaemia; mitochondria; nitric oxide; reactive oxygen species; reperfusion.
© 2020 The Authors. Journal of Cellular and Molecular Medicine published by Foundation for Cellular and Molecular Medicine and John Wiley & Sons Ltd.
Conflict of interest statement
PF is a founder and CEO of Pharmahungary Group, a group of R&D companies.
Figures
References
-
- Sies H.Oxidative stress: introduction In: Sies H. ed. Oxidative Stress: Oxidants and Antioxidants. London: Academic Press, 1991:xv‐xxii.
-
- Dalle‐Donne I, Rossi R, Colombo R, et al. Biomarkers of oxidative damage in human disease. Clin Chem. 2006;52:601‐623. - PubMed
-
- Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. 2008;101:14D‐9D. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
