

Split & mix assembly of DNA libraries for ultrahigh throughput on-bead screening of functional proteins
- PMID: 32383757
- PMCID: PMC7293038
- DOI: 10.1093/nar/gkaa270
Split & mix assembly of DNA libraries for ultrahigh throughput on-bead screening of functional proteins
Abstract
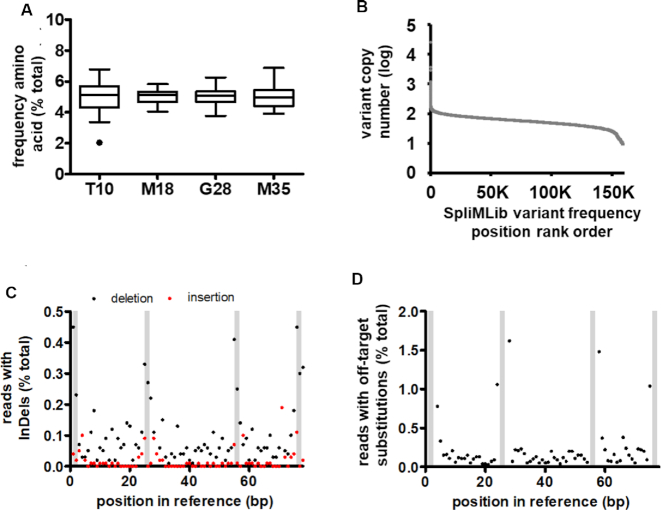
Site-saturation libraries reduce protein screening effort in directed evolution campaigns by focusing on a limited number of rationally chosen residues. However, uneven library synthesis efficiency leads to amino acid bias, remedied at high cost by expensive custom synthesis of oligonucleotides, or through use of proprietary library synthesis platforms. To address these shortcomings, we have devised a method where DNA libraries are constructed on the surface of microbeads by ligating dsDNA fragments onto growing, surface-immobilised DNA, in iterative split-and-mix cycles. This method-termed SpliMLiB for Split-and-Mix Library on Beads-was applied towards the directed evolution of an anti-IgE Affibody (ZIgE), generating a 160,000-membered, 4-site, saturation library on the surface of 8 million monoclonal beads. Deep sequencing confirmed excellent library balance (5.1% ± 0.77 per amino acid) and coverage (99.3%). As SpliMLiB beads are monoclonal, they were amenable to direct functional screening in water-in-oil emulsion droplets with cell-free expression. A FACS-based sorting of the library beads allowed recovery of hits improved in Kd over wild-type ZIgE by up to 3.5-fold, while a consensus mutant of the best hits provided a 10-fold improvement. With SpliMLiB, directed evolution workflows are accelerated by integrating high-quality DNA library generation with an ultra-high throughput protein screening platform.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures




References
-
- Acevedo-Rocha C.G., Hoebenreich S., Reetz M.T.. Iterative saturation mutagenesis: a powerful approach to engineer proteins by systematically simulating Darwinian evolution. Methods Mol. Biol. 2014; 1179:103–128. - PubMed
-
- Kelly R.M., Leemhuis H., Dijkhuizen L.. Conversion of a cyclodextrin glucanotransferase into an alpha-amylase: assessment of directed evolution strategies. Biochemistry. 2007; 46:11216–11222. - PubMed
-
- Parra L.P., Agudo R., Reetz M.T.. Directed evolution by using iterative saturation mutagenesis based on multiresidue sites. ChemBioChem. 2013; 14:2301–2309. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
