

Spider venom-derived peptide induces hyperalgesia in Nav1.7 knockout mice by activating Nav1.9 channels
- PMID: 32385249
- PMCID: PMC7210961
- DOI: 10.1038/s41467-020-16210-y
Spider venom-derived peptide induces hyperalgesia in Nav1.7 knockout mice by activating Nav1.9 channels
Abstract
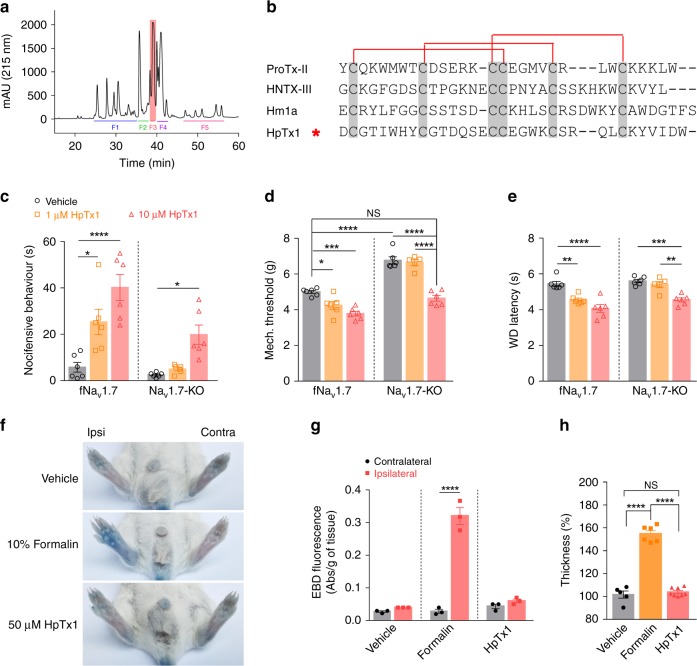
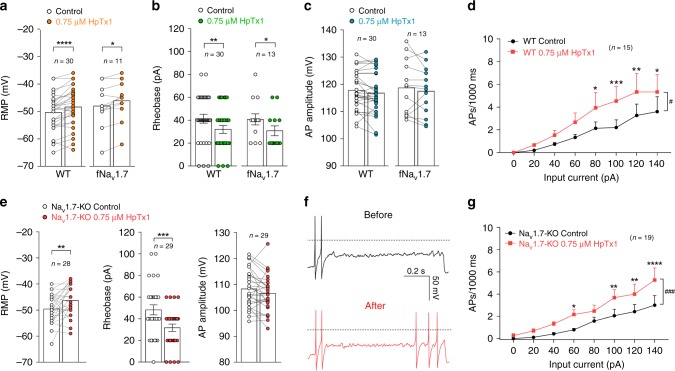
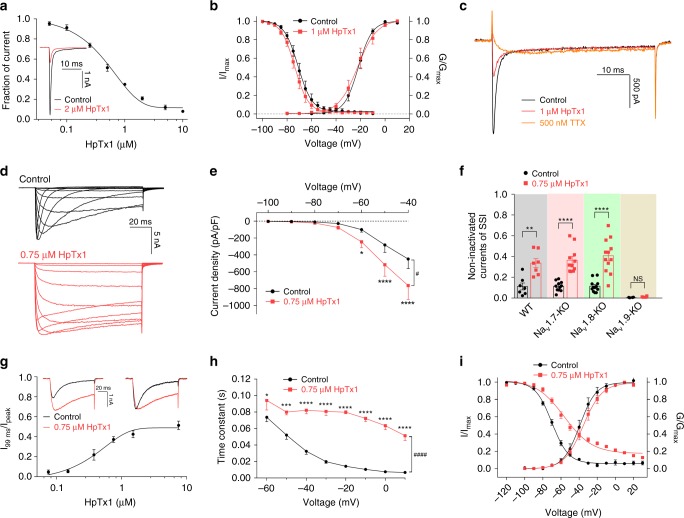
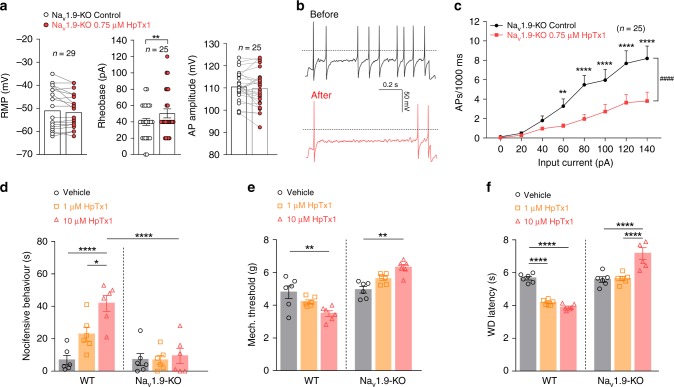
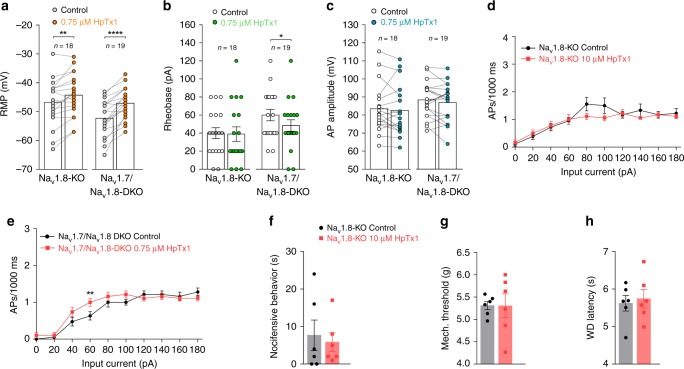
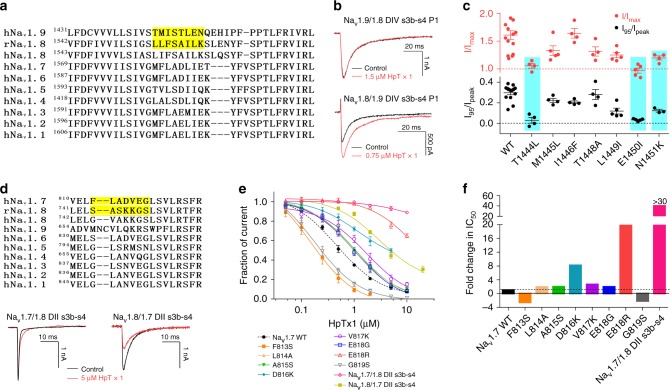
The sodium channels Nav1.7, Nav1.8 and Nav1.9 are critical for pain perception in peripheral nociceptors. Loss of function of Nav1.7 leads to congenital insensitivity to pain in humans. Here we show that the spider peptide toxin called HpTx1, first identified as an inhibitor of Kv4.2, restores nociception in Nav1.7 knockout (Nav1.7-KO) mice by enhancing the excitability of dorsal root ganglion neurons. HpTx1 inhibits Nav1.7 and activates Nav1.9 but does not affect Nav1.8. This toxin produces pain in wild-type (WT) and Nav1.7-KO mice, and attenuates nociception in Nav1.9-KO mice, but has no effect in Nav1.8-KO mice. These data indicate that HpTx1-induced hypersensitivity is mediated by Nav1.9 activation and offers pharmacological insight into the relationship of the three Nav channels in pain signalling.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
