

Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma
- PMID: 32385277
- PMCID: PMC7210975
- DOI: 10.1038/s41467-020-16164-1
Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma
Abstract
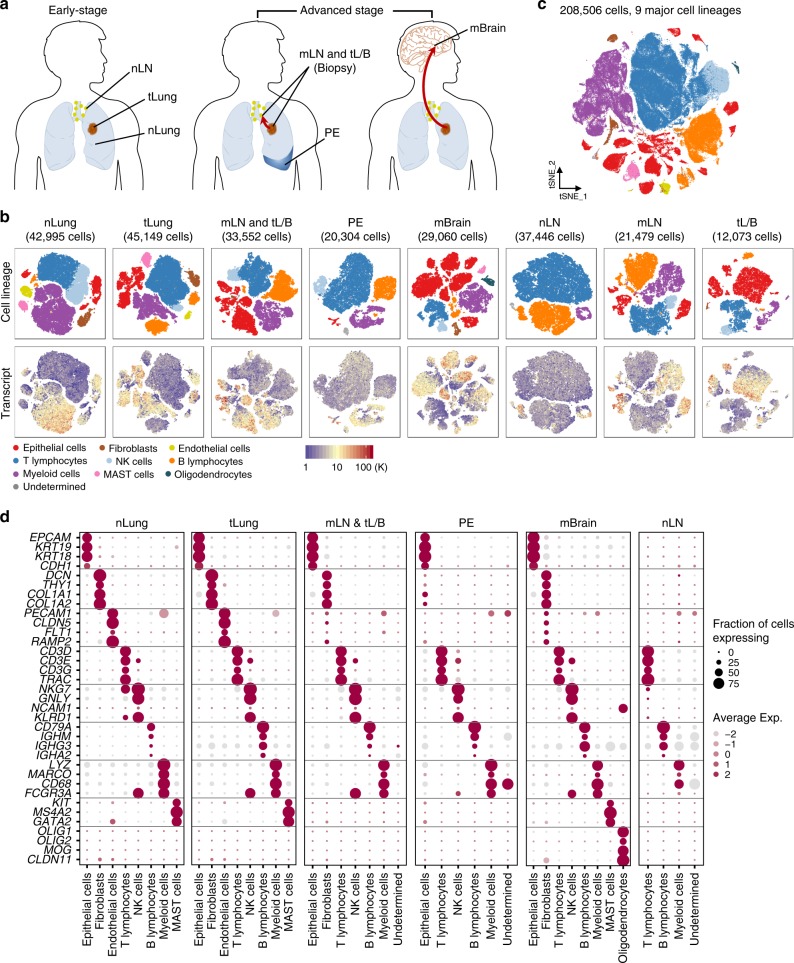
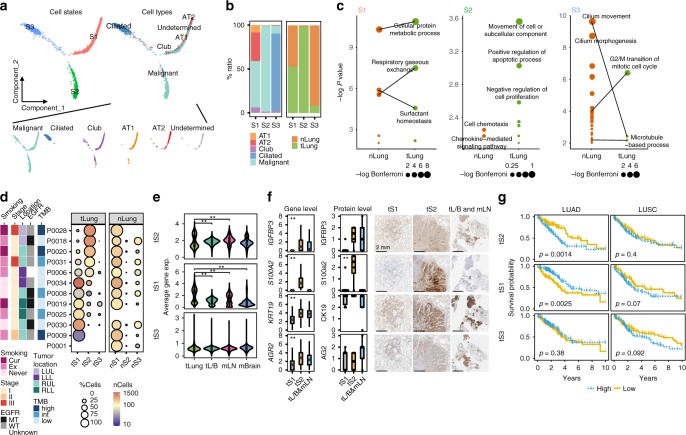
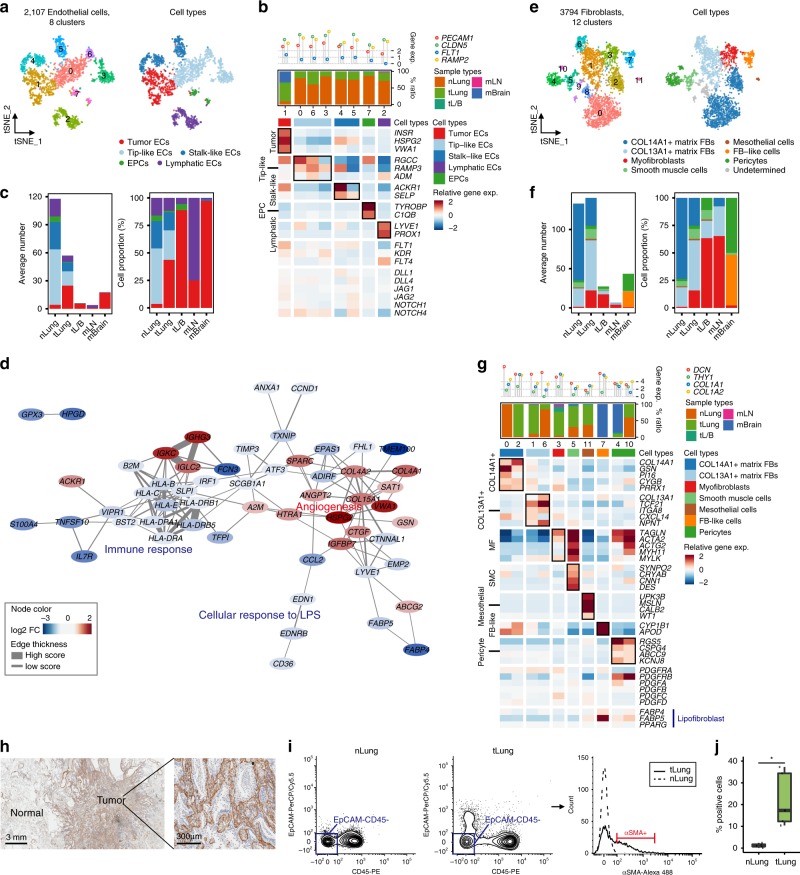
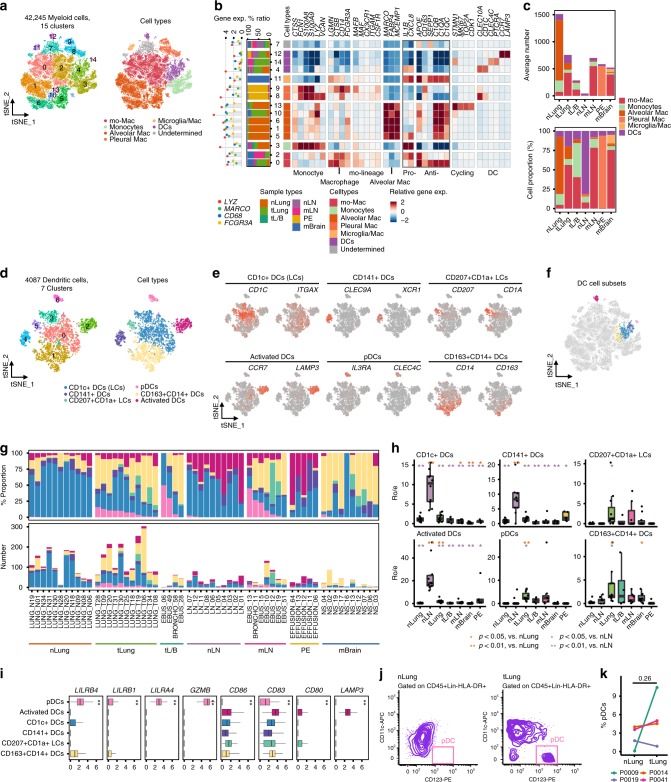
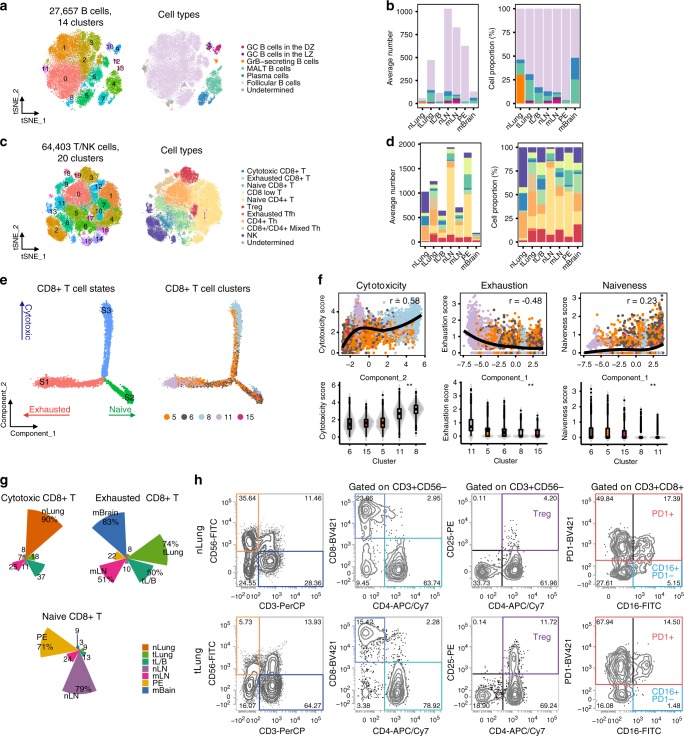
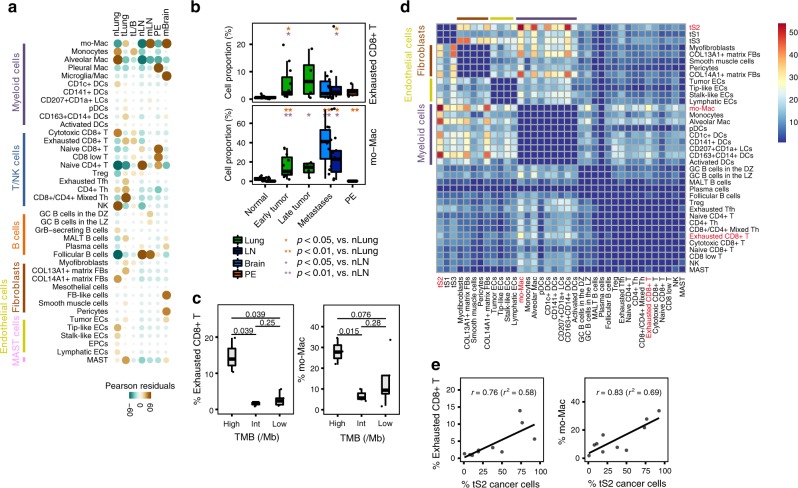
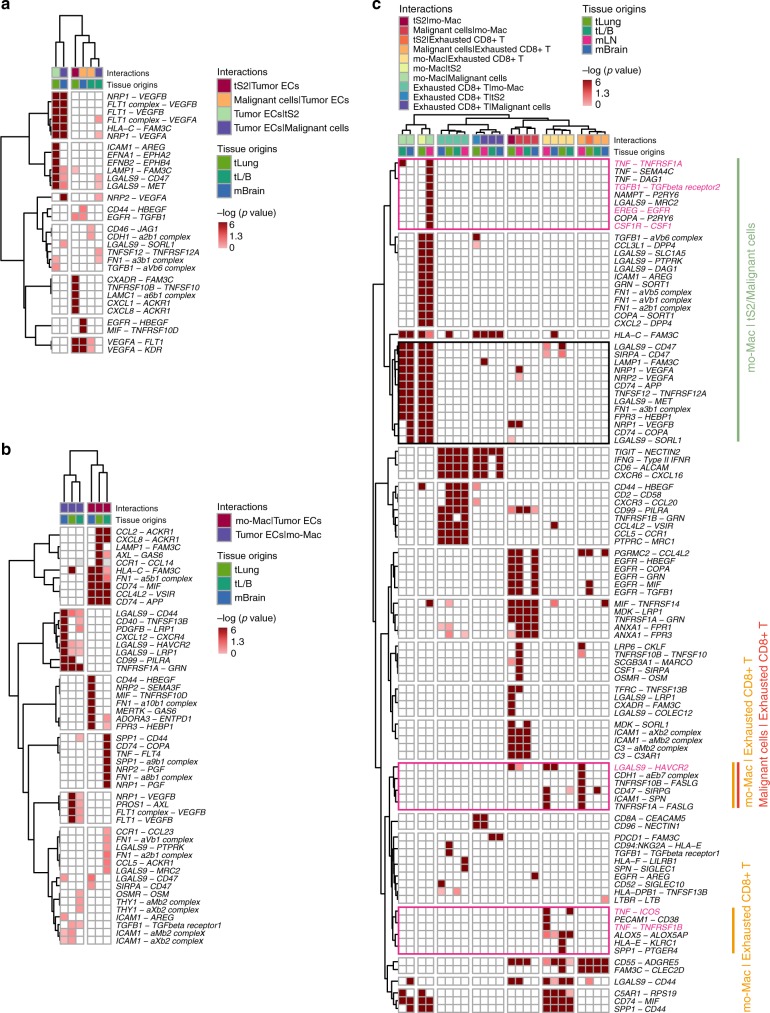
Advanced metastatic cancer poses utmost clinical challenges and may present molecular and cellular features distinct from an early-stage cancer. Herein, we present single-cell transcriptome profiling of metastatic lung adenocarcinoma, the most prevalent histological lung cancer type diagnosed at stage IV in over 40% of all cases. From 208,506 cells populating the normal tissues or early to metastatic stage cancer in 44 patients, we identify a cancer cell subtype deviating from the normal differentiation trajectory and dominating the metastatic stage. In all stages, the stromal and immune cell dynamics reveal ontological and functional changes that create a pro-tumoral and immunosuppressive microenvironment. Normal resident myeloid cell populations are gradually replaced with monocyte-derived macrophages and dendritic cells, along with T-cell exhaustion. This extensive single-cell analysis enhances our understanding of molecular and cellular dynamics in metastatic lung cancer and reveals potential diagnostic and therapeutic targets in cancer-microenvironment interactions.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Comprehensive analyses for the coagulation and macrophage-related genes to reveal their joint roles in the prognosis and immunotherapy of lung adenocarcinoma patients.Front Immunol. 2023 Oct 31;14:1273422. doi: 10.3389/fimmu.2023.1273422. eCollection 2023. Front Immunol. 2023. PMID: 38022584 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
