

Coding potential and sequence conservation of SARS-CoV-2 and related animal viruses
- PMID: 32387562
- PMCID: PMC7199688
- DOI: 10.1016/j.meegid.2020.104353
Coding potential and sequence conservation of SARS-CoV-2 and related animal viruses
Abstract
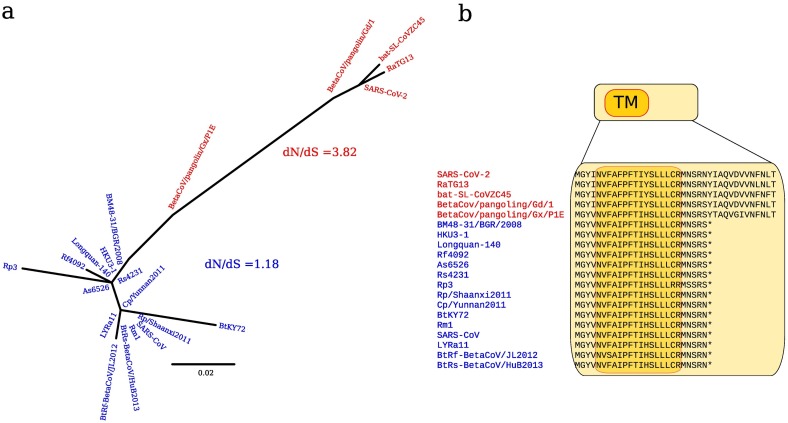
In December 2019, a novel human-infecting coronavirus (SARS-CoV-2) was recognized in China. In a few months, SARS-CoV-2 has caused thousands of disease cases and deaths in several countries. Phylogenetic analyses indicated that SARS-CoV-2 clusters with SARS-CoV in the Sarbecovirus subgenus and viruses related to SARS-CoV-2 were identified from bats and pangolins. Coronaviruses have long and complex genomes with high plasticity in terms of gene content. To date, the coding potential of SARS-CoV-2 remains partially unknown. We thus used available sequences of bat and pangolin viruses to determine the selective events that shaped the genome structure of SARS-CoV-2 and to assess its coding potential. By searching for signals of significantly reduced variability at synonymous sites (dS), we identified six genomic regions, one of these corresponding to the programmed -1 ribosomal frameshift. The most prominent signal of dS reduction was observed within the E gene. A genome-wide analysis of conserved RNA structures indicated that this region harbors a putative functional RNA element that is shared with the SARS-CoV lineage. Additional signals of reduced dS indicated the presence of internal ORFs. Whereas the presence ORF9a (internal to N) was previously proposed by homology with a well characterized protein of SARS-CoV, ORF3h (for hypothetical, within ORF3a) was not previously described. The predicted product of ORF3h has 90% identity with the corresponding predicted product of SARS-CoV and displays features suggestive of a viroporin. Finally, analysis of the putative ORF10 revealed high dN/dS (3.82) in SARS-CoV-2 and related coronaviruses. In the SARS-CoV lineage, the ORF is predicted to encode a truncated protein and is neutrally evolving. These data suggest that ORF10 encodes a functional protein in SARS-CoV-2 and that positive selection is driving its evolution. Experimental analyses will be necessary to validate and characterize the coding and non-coding functional elements we identified.
Keywords: Coding potential; Coronaviruses; Functional RNA elements; SARS-CoV-2.
Copyright © 2020 Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
