

Emergence of genomic diversity and recurrent mutations in SARS-CoV-2
- PMID: 32387564
- PMCID: PMC7199730
- DOI: 10.1016/j.meegid.2020.104351
Emergence of genomic diversity and recurrent mutations in SARS-CoV-2
Abstract
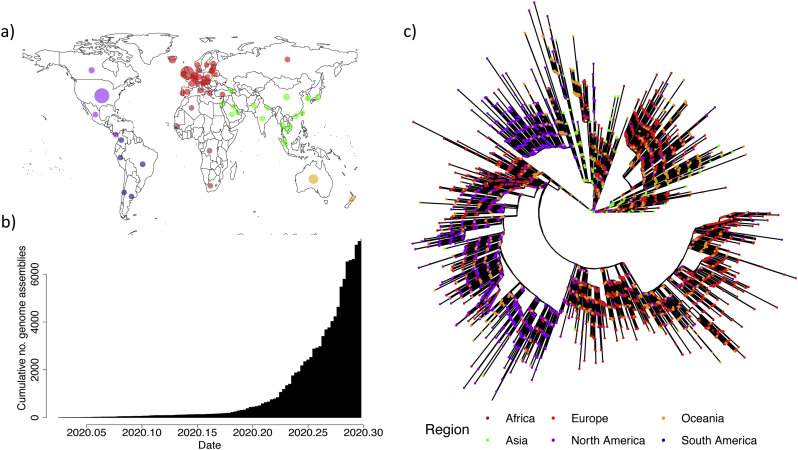
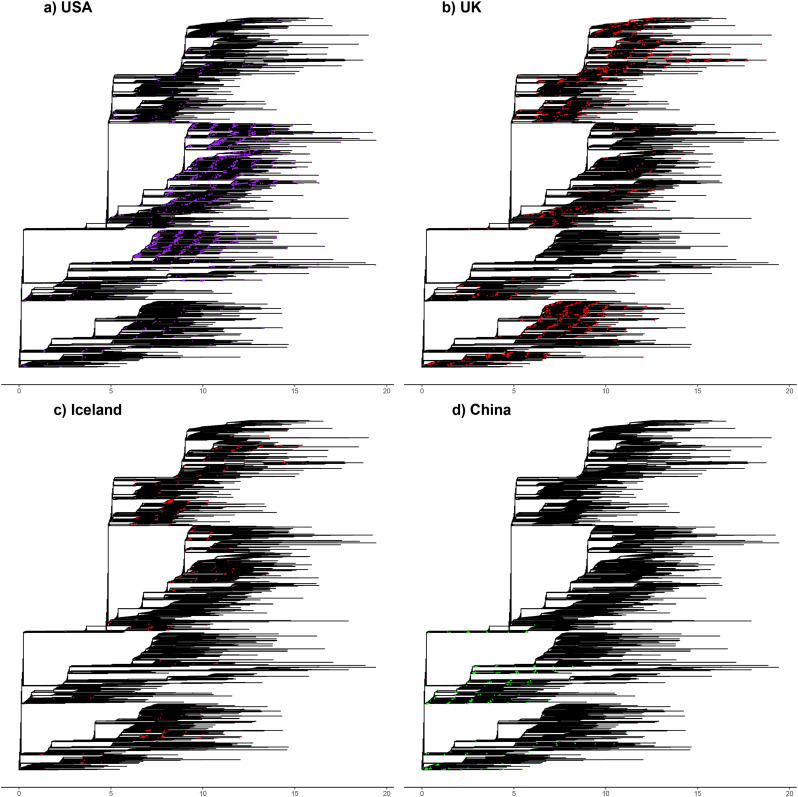
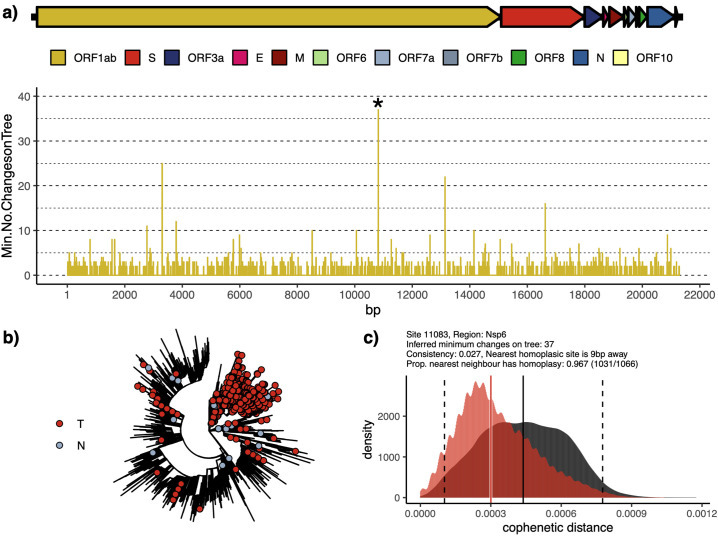
SARS-CoV-2 is a SARS-like coronavirus of likely zoonotic origin first identified in December 2019 in Wuhan, the capital of China's Hubei province. The virus has since spread globally, resulting in the currently ongoing COVID-19 pandemic. The first whole genome sequence was published on January 5 2020, and thousands of genomes have been sequenced since this date. This resource allows unprecedented insights into the past demography of SARS-CoV-2 but also monitoring of how the virus is adapting to its novel human host, providing information to direct drug and vaccine design. We curated a dataset of 7666 public genome assemblies and analysed the emergence of genomic diversity over time. Our results are in line with previous estimates and point to all sequences sharing a common ancestor towards the end of 2019, supporting this as the period when SARS-CoV-2 jumped into its human host. Due to extensive transmission, the genetic diversity of the virus in several countries recapitulates a large fraction of its worldwide genetic diversity. We identify regions of the SARS-CoV-2 genome that have remained largely invariant to date, and others that have already accumulated diversity. By focusing on mutations which have emerged independently multiple times (homoplasies), we identify 198 filtered recurrent mutations in the SARS-CoV-2 genome. Nearly 80% of the recurrent mutations produced non-synonymous changes at the protein level, suggesting possible ongoing adaptation of SARS-CoV-2. Three sites in Orf1ab in the regions encoding Nsp6, Nsp11, Nsp13, and one in the Spike protein are characterised by a particularly large number of recurrent mutations (>15 events) which may signpost convergent evolution and are of particular interest in the context of adaptation of SARS-CoV-2 to the human host. We additionally provide an interactive user-friendly web-application to query the alignment of the 7666 SARS-CoV-2 genomes.
Keywords: Betacoronavirus; Homoplasies; Mutation; Phylogenetics.
Copyright © 2020 Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors have no competing interests to declare.
Figures
References
-
- Anderson R.M., May R.M. Oxford University Press; Oxford: 1991. Infectious Diseases of Humans. Dynamics and Control.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
