

TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer's Disease
- PMID: 32391019
- PMCID: PMC7190872
- DOI: 10.3389/fimmu.2020.00724
TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer's Disease
Abstract
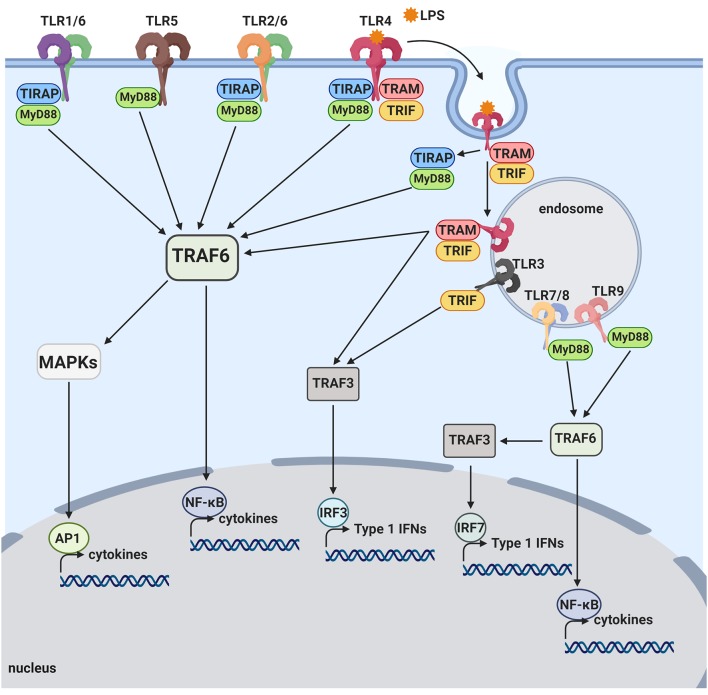
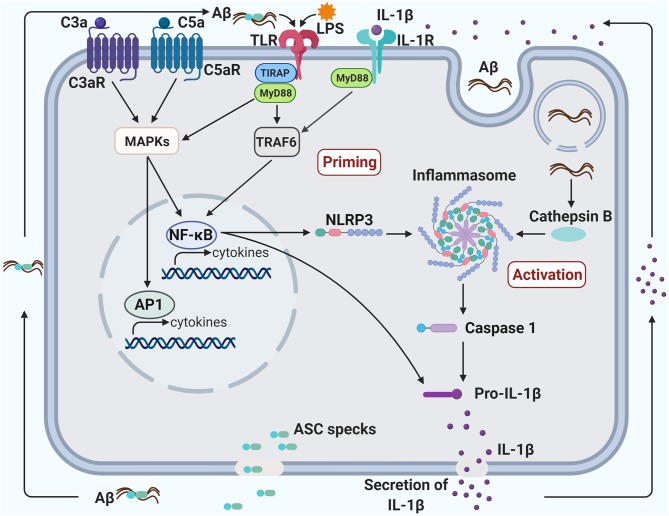
Amyloid plaques, mainly composed of abnormally aggregated amyloid β-protein (Aβ) in the brain parenchyma, and neurofibrillary tangles (NFTs), consisting of hyperphosphorylated tau protein aggregates in neurons, are two pathological hallmarks of Alzheimer's disease (AD). Aβ fibrils and tau aggregates in the brain are closely associated with neuroinflammation and synapse loss, characterized by activated microglia and dystrophic neurites. Genome-wide genetic association studies revealed important roles of innate immune cells in the pathogenesis of late-onset AD by recognizing a dozen genetic risk loci that modulate innate immune activities. Furthermore, microglia, brain resident innate immune cells, have been increasingly recognized to play key, opposing roles in AD pathogenesis by either eliminating toxic Aβ aggregates and enhancing neuronal plasticity or producing proinflammatory cytokines, reactive oxygen species, and synaptotoxicity. Aggregated Aβ binds to toll-like receptor 4 (TLR4) and activates microglia, resulting in increased phagocytosis and cytokine production. Complement components are associated with amyloid plaques and NFTs. Aggregated Aβ can activate complement, leading to synapse pruning and loss by microglial phagocytosis. Systemic inflammation can activate microglial TLR4, NLRP3 inflammasome, and complement in the brain, leading to neuroinflammation, Aβ accumulation, synapse loss and neurodegeneration. The host immune response has been shown to function through complex crosstalk between the TLR, complement and inflammasome signaling pathways. Accordingly, targeting the molecular mechanisms underlying the TLR-complement-NLRP3 inflammasome signaling pathways can be a preventive and therapeutic approach for AD.
Keywords: Alzheimer's disease; TLR4; amyloid; complement; inflammasome; synapse.
Copyright © 2020 Yang, Wise and Fukuchi.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical