

Glycogen synthase kinase-3 inhibition overcomes epithelial-mesenchymal transition-associated resistance to osimertinib in EGFR-mutant lung cancer
- PMID: 32391602
- PMCID: PMC7385349
- DOI: 10.1111/cas.14454
Glycogen synthase kinase-3 inhibition overcomes epithelial-mesenchymal transition-associated resistance to osimertinib in EGFR-mutant lung cancer
Abstract
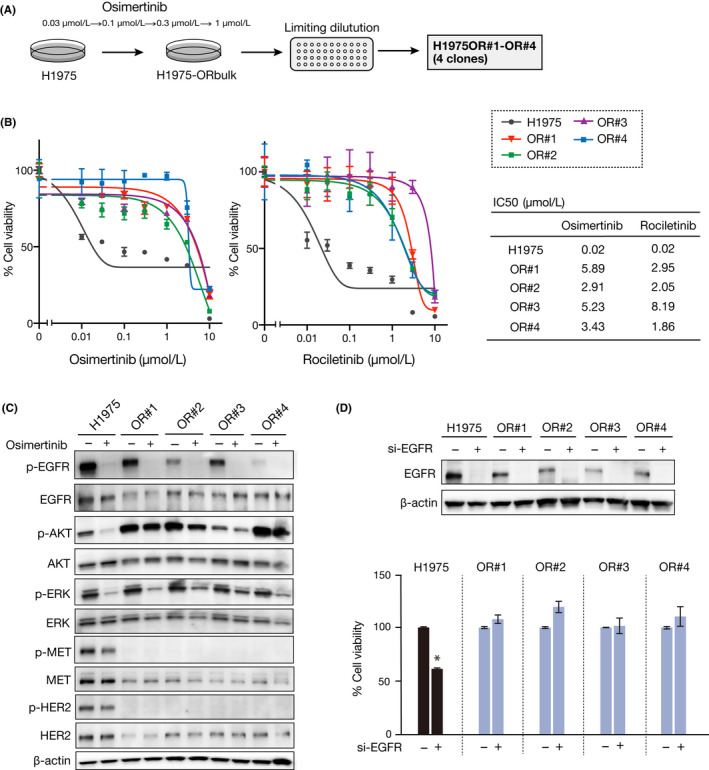
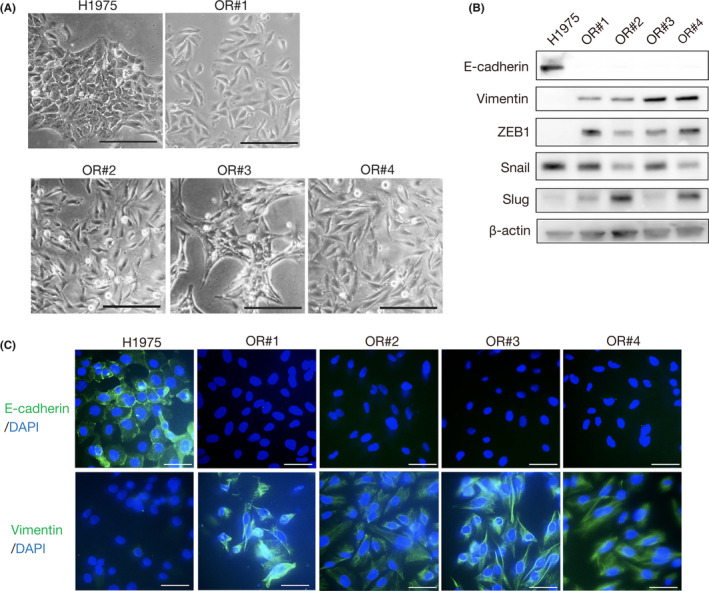
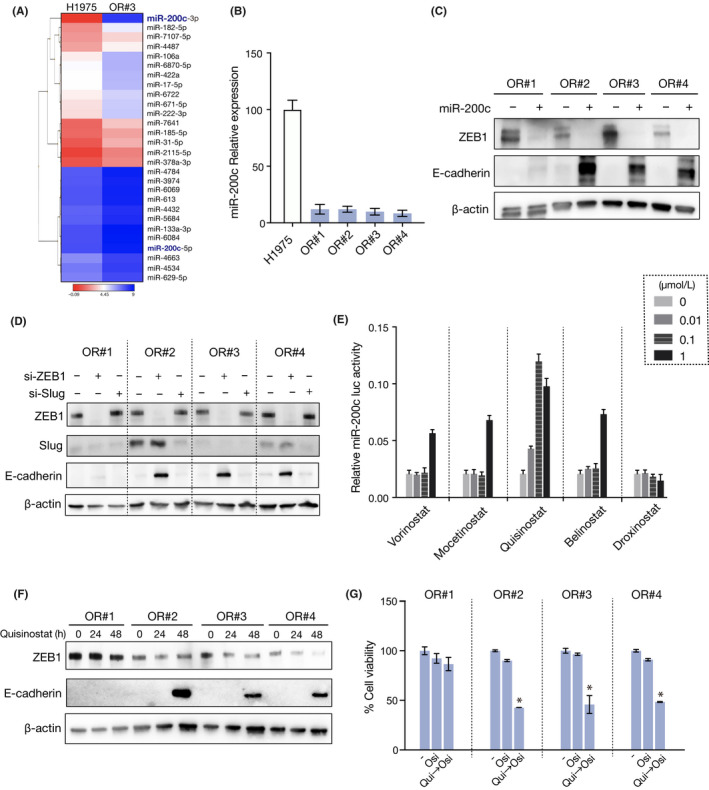
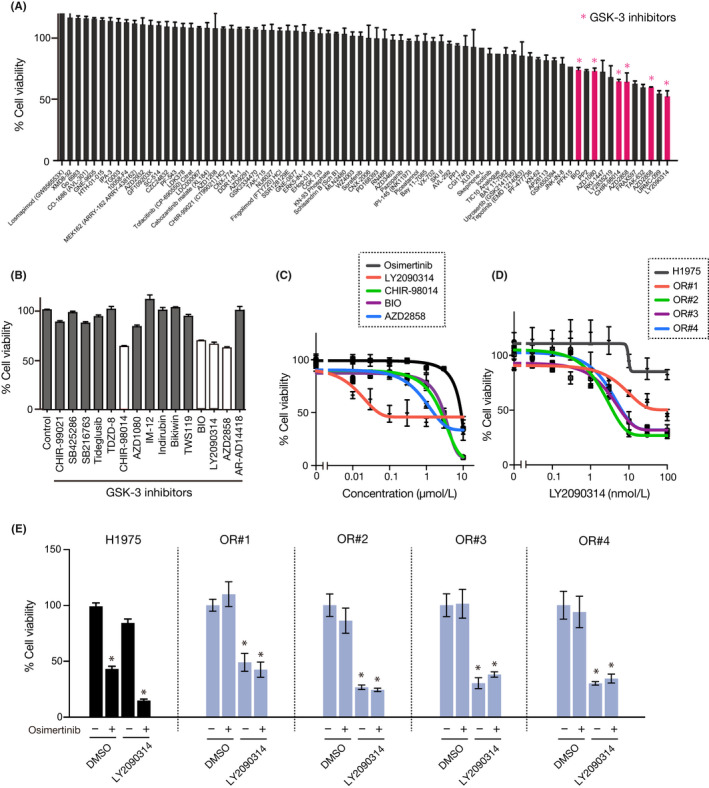
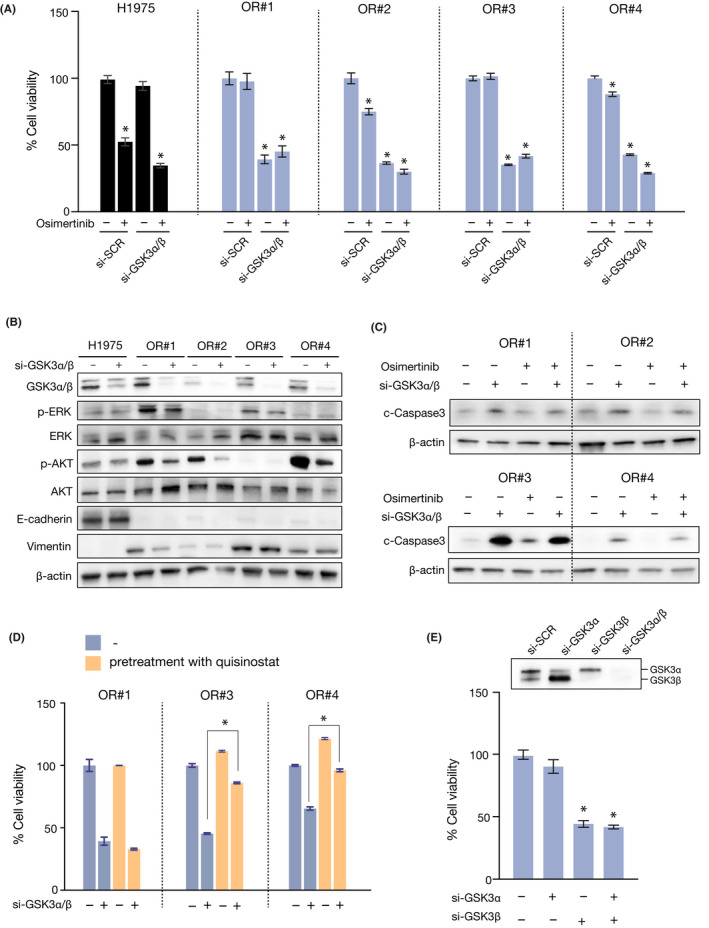
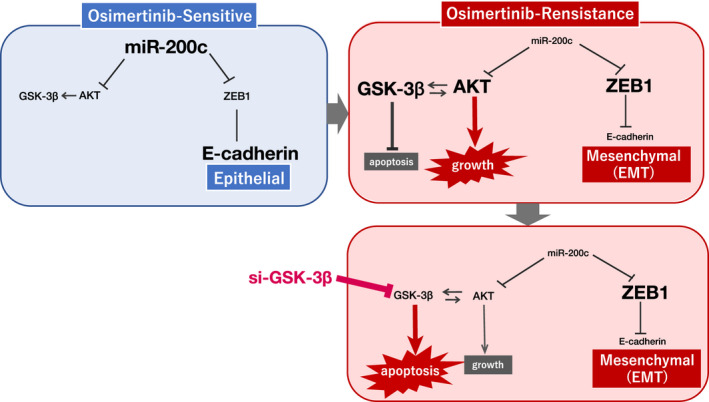
A novel epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor, osimertinib, has marked efficacy in patients with EGFR-mutant lung cancer. While epithelial-mesenchymal transition (EMT) plays a role in the resistance to various targeted drugs, its involvement in EGFR-inhibitor resistance remains largely unknown. Preclinical experiments with osimertinib-resistant lung cancer cells showed that EMT was associated with decreased microRNA-200c and increased ZEB1 expression. In several resistant clone cells, pretreatment with the histone deacetylase inhibitor quisinostat helped overcome the resistance by reverting EMT. Furthermore, drug screening from a library of 100 kinase inhibitors indicated that Glycogen synthase kinase-3 (GSK-3) inhibitors, such as LY2090314, markedly inhibited the growth and induced apoptosis of resistant cells, specifically those with a mesenchymal phenotype. These results suggest that GSK-3 inhibition could be useful to circumvent EMT-associated resistance to osimertinib in EGFR-mutant lung cancer.
Keywords: epidermal growth factor receptor; epithelial-mesenchymal transition; glycogen synthase kinase-3; osimertinib; resistance.
© 2020 The Authors. Cancer Science published by John Wiley & Sons Australia, Ltd on behalf of Japanese Cancer Association.
Conflict of interest statement
Seiji Yano obtained speaker’s fees from AstraZeneca, Chugai Pharma, Boehringer‐Ingelheim Japan, Novartis, and Pfizer, and research grants from Chugai Pharma, Boehringer‐Ingelheim Japan, and Novartis. The other authors have nothing to disclose.
Figures
References
-
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380‐2388. - PubMed
-
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 2012;13:239‐246. - PubMed
-
- Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2005;352:786‐792. - PubMed
-
- Finlay MR, Anderton M, Ashton S, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57:8249‐8267. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
