

Developmental trajectory of prehematopoietic stem cell formation from endothelium
- PMID: 32392346
- PMCID: PMC7426642
- DOI: 10.1182/blood.2020004801
Developmental trajectory of prehematopoietic stem cell formation from endothelium
Abstract
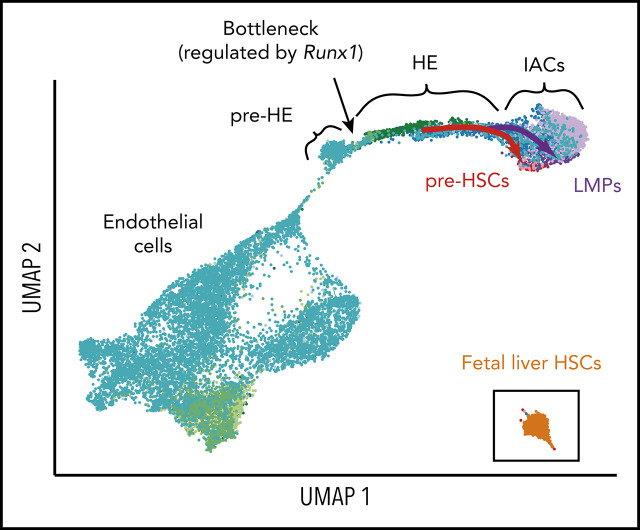
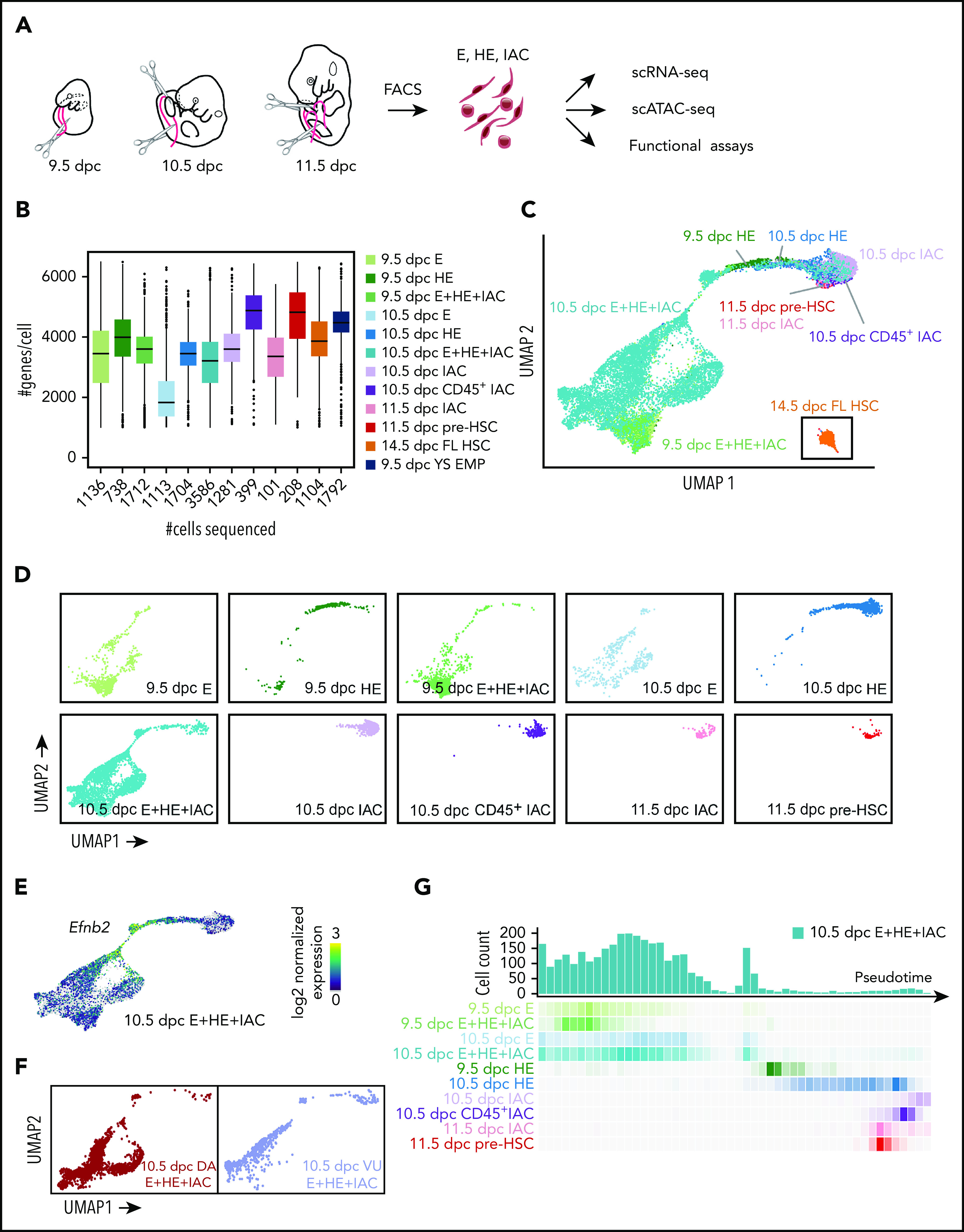
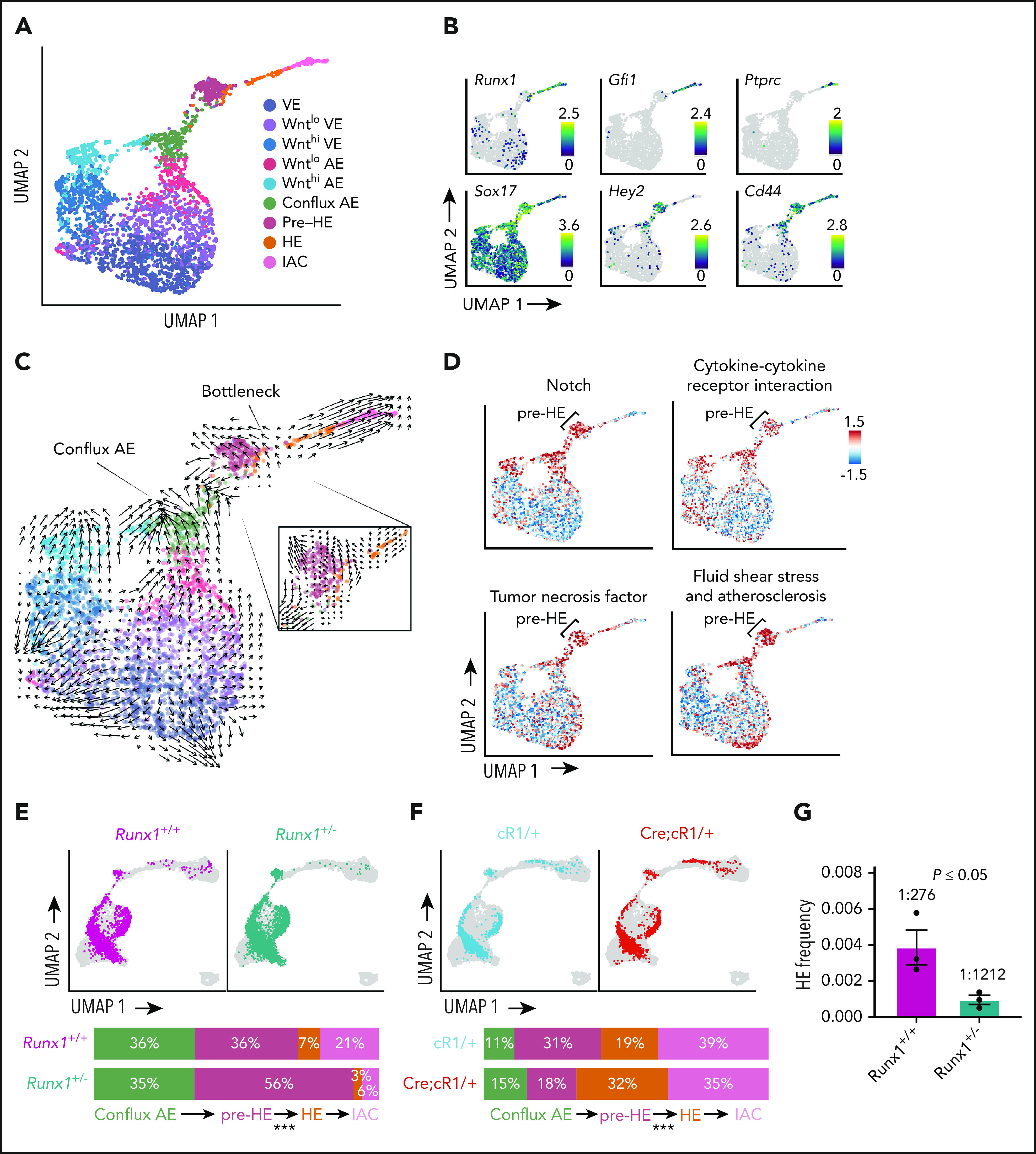
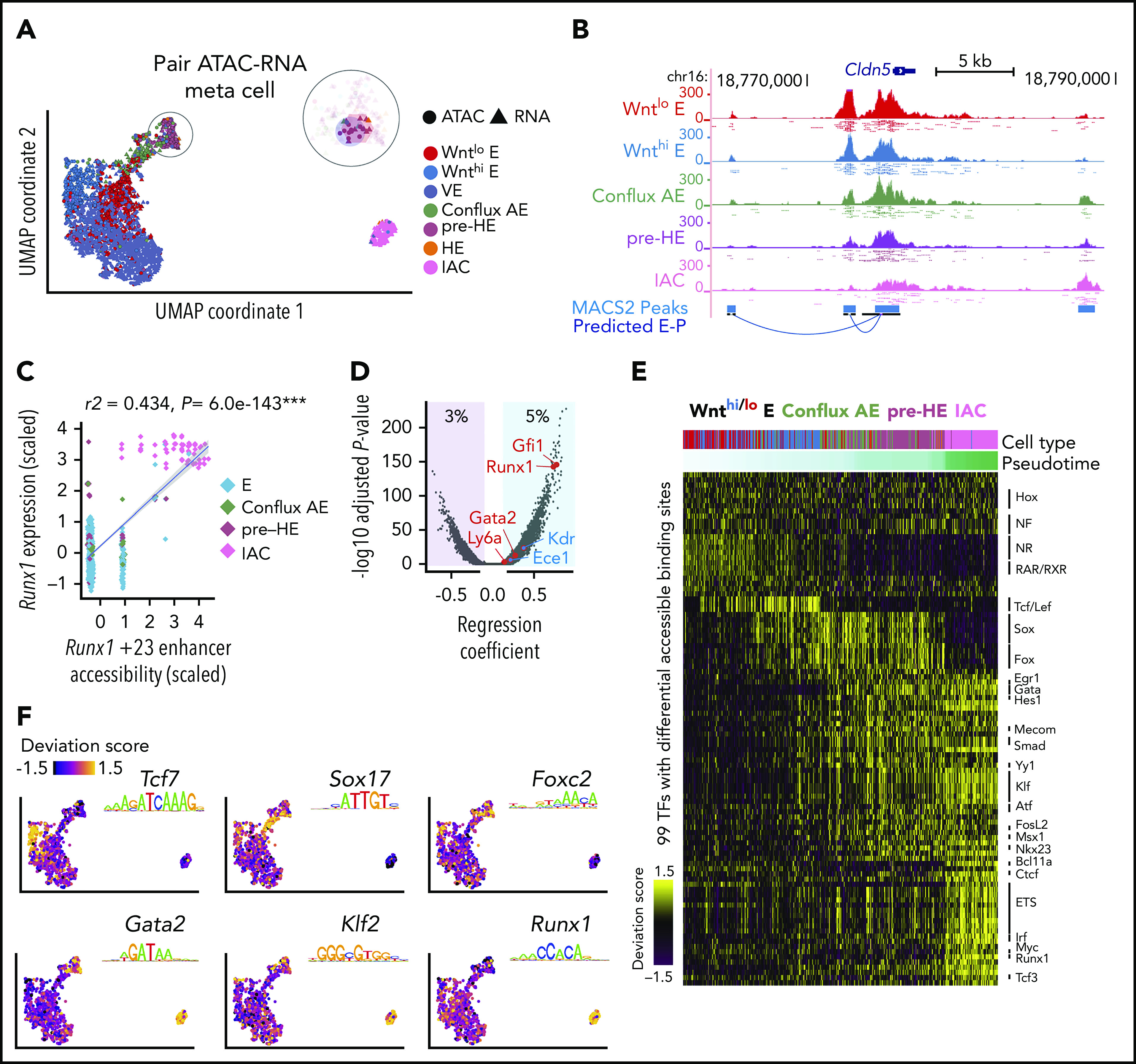
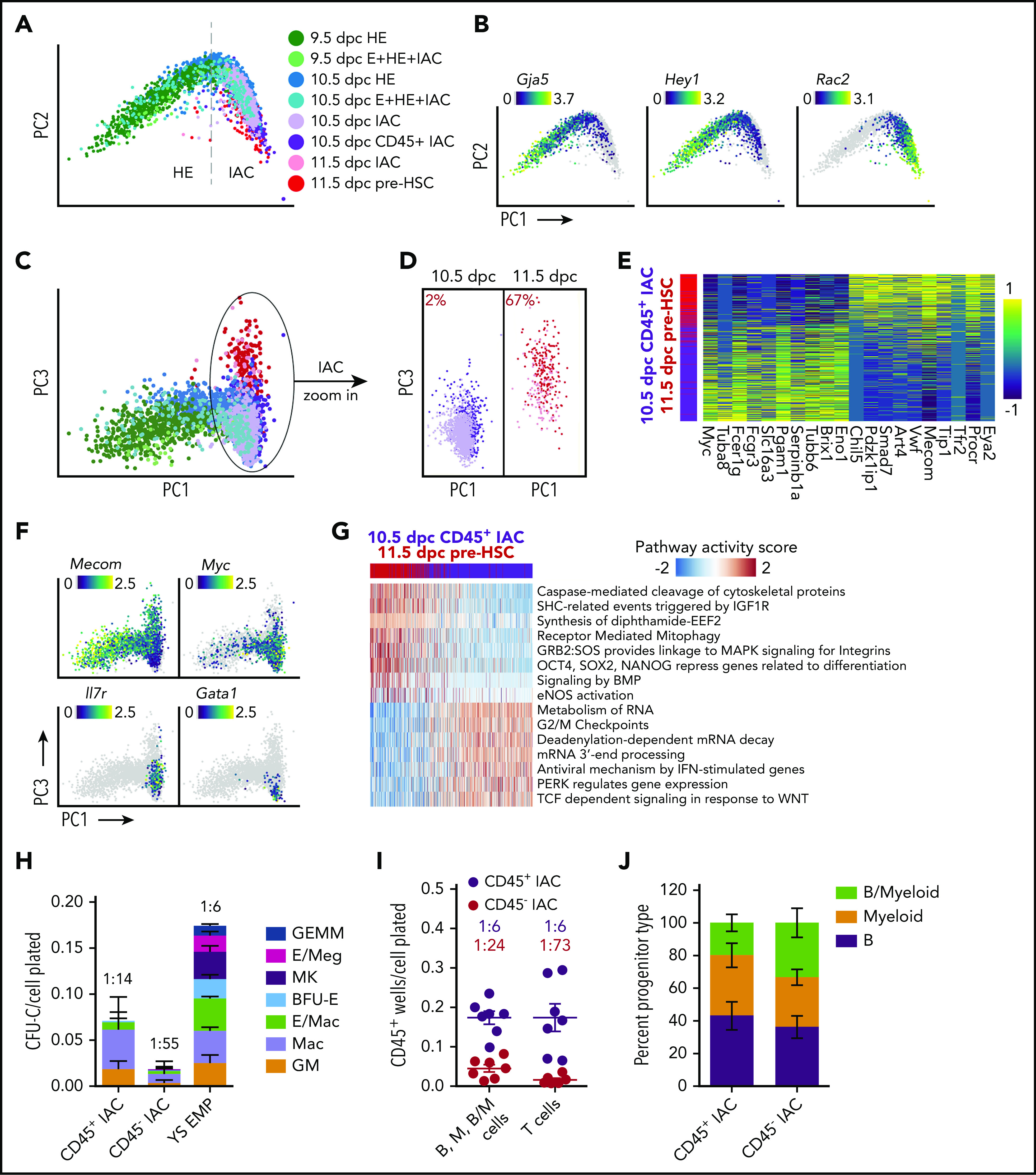
Hematopoietic stem and progenitor cells (HSPCs) in the bone marrow are derived from a small population of hemogenic endothelial (HE) cells located in the major arteries of the mammalian embryo. HE cells undergo an endothelial to hematopoietic cell transition, giving rise to HSPCs that accumulate in intra-arterial clusters (IAC) before colonizing the fetal liver. To examine the cell and molecular transitions between endothelial (E), HE, and IAC cells, and the heterogeneity of HSPCs within IACs, we profiled ∼40 000 cells from the caudal arteries (dorsal aorta, umbilical, vitelline) of 9.5 days post coitus (dpc) to 11.5 dpc mouse embryos by single-cell RNA sequencing and single-cell assay for transposase-accessible chromatin sequencing. We identified a continuous developmental trajectory from E to HE to IAC cells, with identifiable intermediate stages. The intermediate stage most proximal to HE, which we term pre-HE, is characterized by increased accessibility of chromatin enriched for SOX, FOX, GATA, and SMAD motifs. A developmental bottleneck separates pre-HE from HE, with RUNX1 dosage regulating the efficiency of the pre-HE to HE transition. A distal candidate Runx1 enhancer exhibits high chromatin accessibility specifically in pre-HE cells at the bottleneck, but loses accessibility thereafter. Distinct developmental trajectories within IAC cells result in 2 populations of CD45+ HSPCs; an initial wave of lymphomyeloid-biased progenitors, followed by precursors of hematopoietic stem cells (pre-HSCs). This multiomics single-cell atlas significantly expands our understanding of pre-HSC ontogeny.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures


Comment in
-
Decoding hematopoietic stem cells' birth.Blood. 2020 Aug 13;136(7):775-776. doi: 10.1182/blood.2020006612. Blood. 2020. PMID: 32790854 No abstract available.
References
-
- Auerbach R, Huang H, Lu L. Hematopoietic stem cells in the mouse embryonic yolk sac. Stem Cells. 1996;14(3):269-280. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
