

New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand?
- PMID: 32392694
- PMCID: PMC7246502
- DOI: 10.3390/ijms21093297
New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand?
Abstract
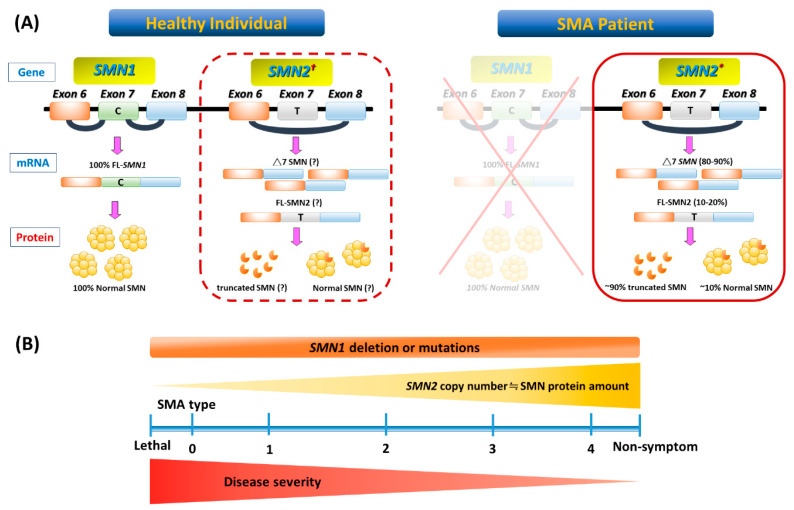
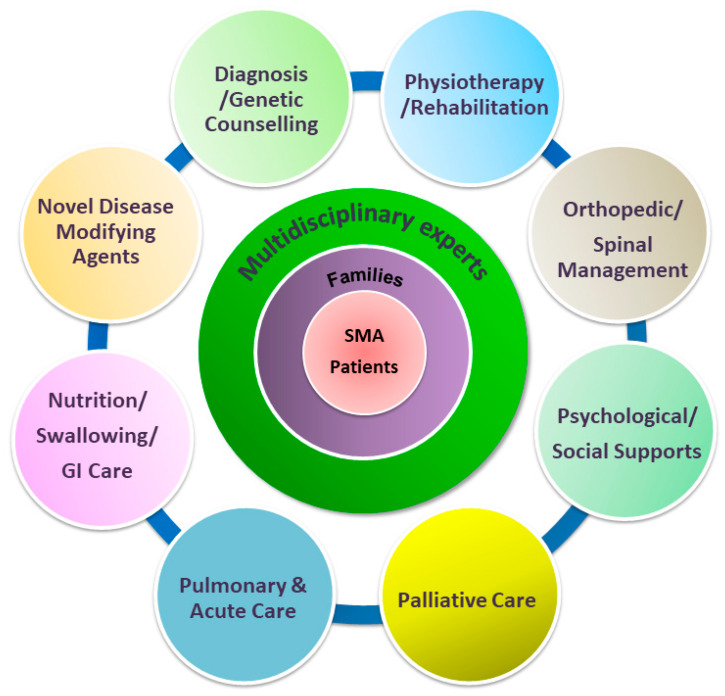
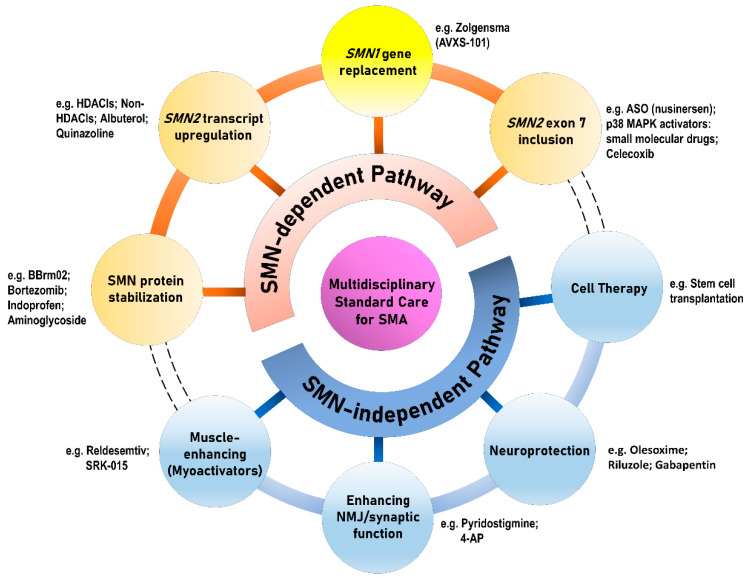
Spinal muscular atrophy (SMA) is a congenital neuromuscular disorder characterized by motor neuron loss, resulting in progressive weakness. SMA is notable in the health care community because it accounts for the most common cause of infant death resulting from a genetic defect. SMA is caused by low levels of the survival motor neuron protein (SMN) resulting from SMN1 gene mutations or deletions. However, patients always harbor various copies of SMN2, an almost identical but functionally deficient copy of the gene. A genotype-phenotype correlation suggests that SMN2 is a potent disease modifier for SMA, which also represents the primary target for potential therapies. Increasing comprehension of SMA pathophysiology, including the characterization of SMN1 and SMN2 genes and SMN protein functions, has led to the development of multiple therapeutic approaches. Until the end of 2016, no cure was available for SMA, and management consisted of supportive measures. Two breakthrough SMN-targeted treatments, either using antisense oligonucleotides (ASOs) or virus-mediated gene therapy, have recently been approved. These two novel therapeutics have a common objective: to increase the production of SMN protein in MNs and thereby improve motor function and survival. However, neither therapy currently provides a complete cure. Treating patients with SMA brings new responsibilities and unique dilemmas. As SMA is such a devastating disease, it is reasonable to assume that a unique therapeutic solution may not be sufficient. Current approaches under clinical investigation differ in administration routes, frequency of dosing, intrathecal versus systemic delivery, and mechanisms of action. Besides, emerging clinical trials evaluating the efficacy of either SMN-dependent or SMN-independent approaches are ongoing. This review aims to address the different knowledge gaps between genotype, phenotypes, and potential therapeutics.
Keywords: clinical care; novel therapy; spinal muscular atrophy; survival motor neuron protein.
Conflict of interest statement
The author declares no conflict of interest.
Figures
References
-
- Crawford T.O., Paushkin S.V., Kobayashi D.T., Forrest S.J., Joyce C.L., Finkel R.S., Kaufmann P., Swoboda K.J., Tiziano D., Lomastro R., et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS ONE. 2012;7:e33572. doi: 10.1371/journal.pone.0033572. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
