

MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data
- PMID: 32393329
- PMCID: PMC7212577
- DOI: 10.1186/s13059-020-02015-1
MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data
Abstract
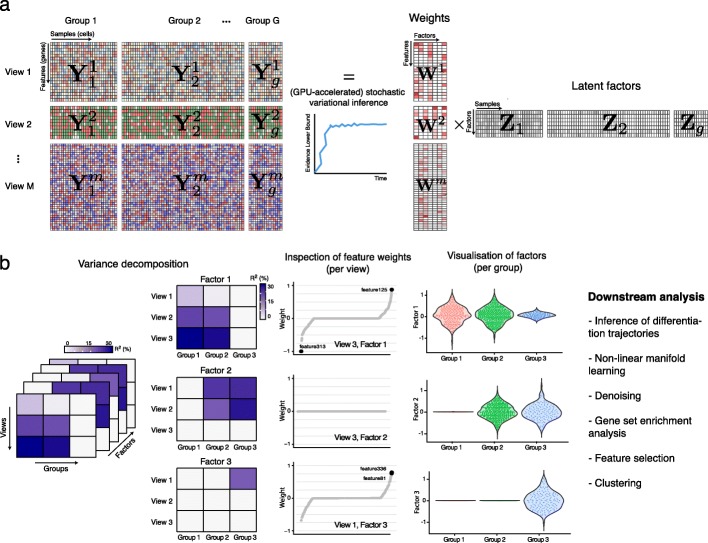
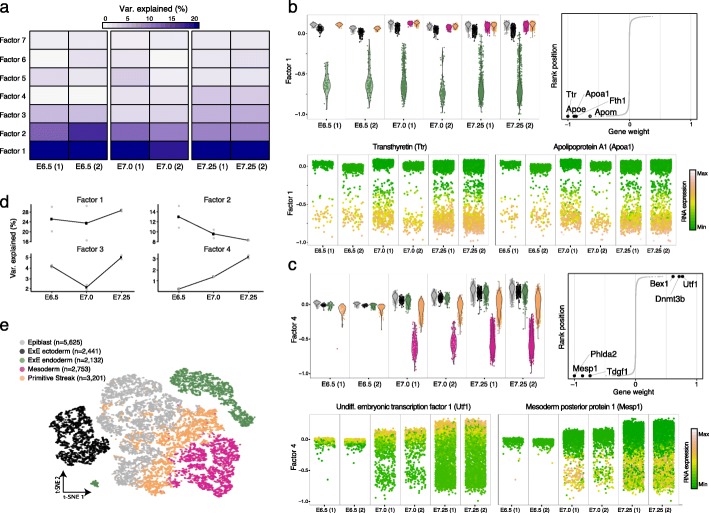
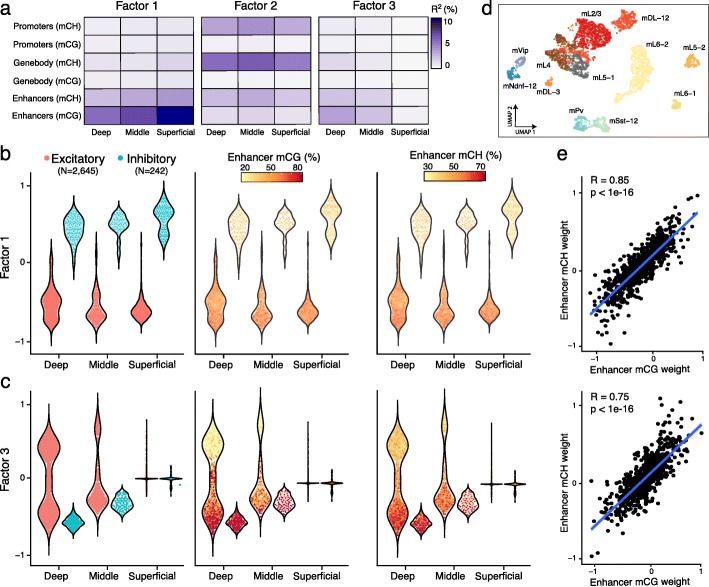
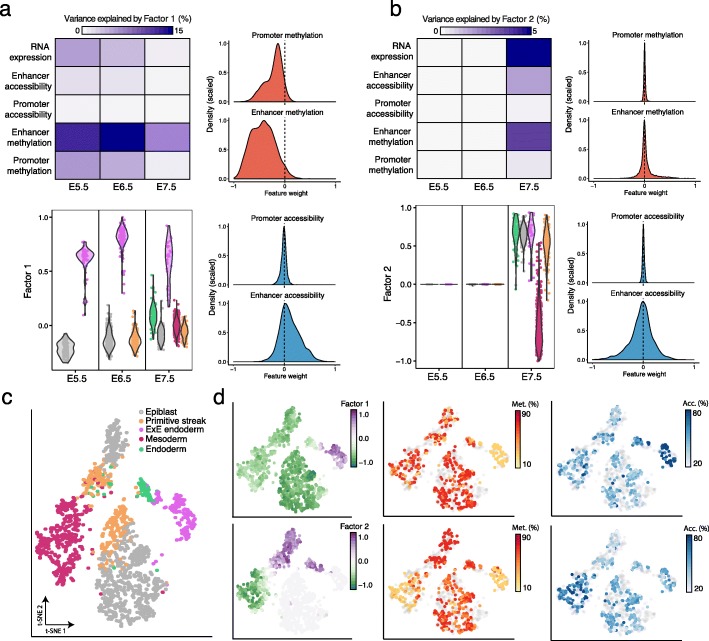
Technological advances have enabled the profiling of multiple molecular layers at single-cell resolution, assaying cells from multiple samples or conditions. Consequently, there is a growing need for computational strategies to analyze data from complex experimental designs that include multiple data modalities and multiple groups of samples. We present Multi-Omics Factor Analysis v2 (MOFA+), a statistical framework for the comprehensive and scalable integration of single-cell multi-modal data. MOFA+ reconstructs a low-dimensional representation of the data using computationally efficient variational inference and supports flexible sparsity constraints, allowing to jointly model variation across multiple sample groups and data modalities.
Keywords: Data integration; Factor analysis; Multi-omics; Single cell.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
