

Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation
- PMID: 32393626
- PMCID: PMC7261084
- DOI: 10.1073/pnas.1911870117
Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation
Erratum in
-
Correction for Luther et al., Hepatic gap junctions amplify alcohol liver injury by propagating cGAS-mediated IRF3 activation.Proc Natl Acad Sci U S A. 2020 Jul 14;117(28):16704. doi: 10.1073/pnas.2010186117. Epub 2020 Jul 6. Proc Natl Acad Sci U S A. 2020. PMID: 32631979 Free PMC article. No abstract available.
Abstract
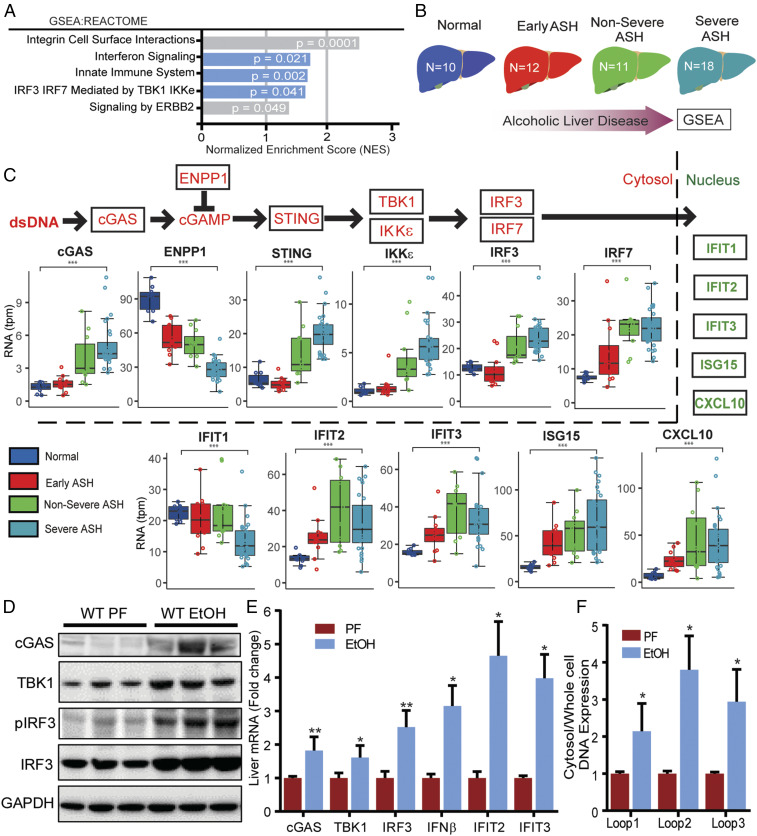
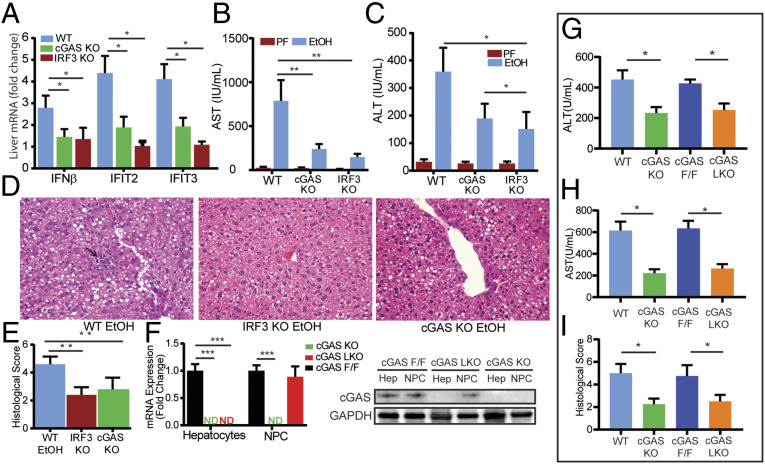
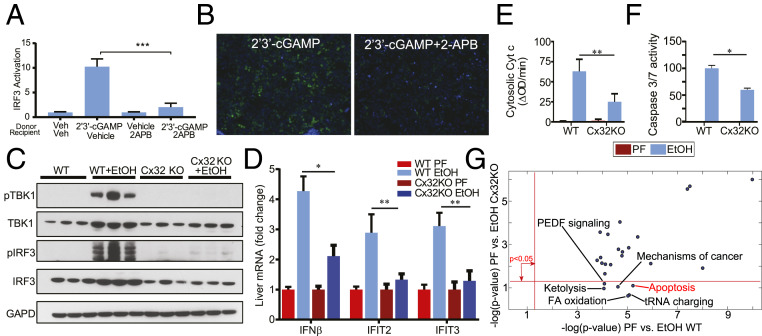
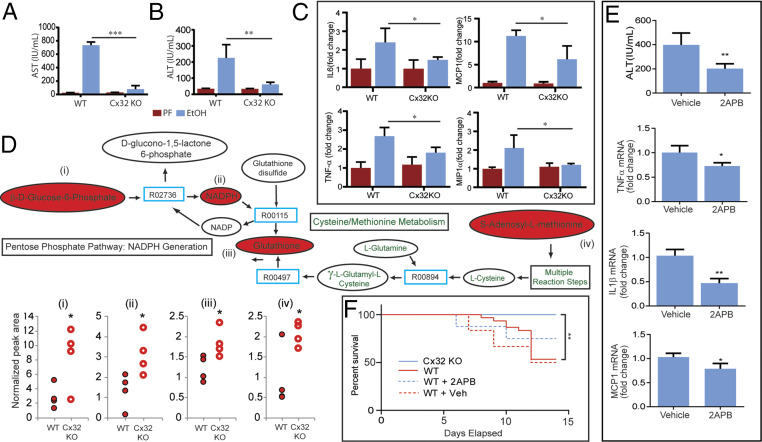
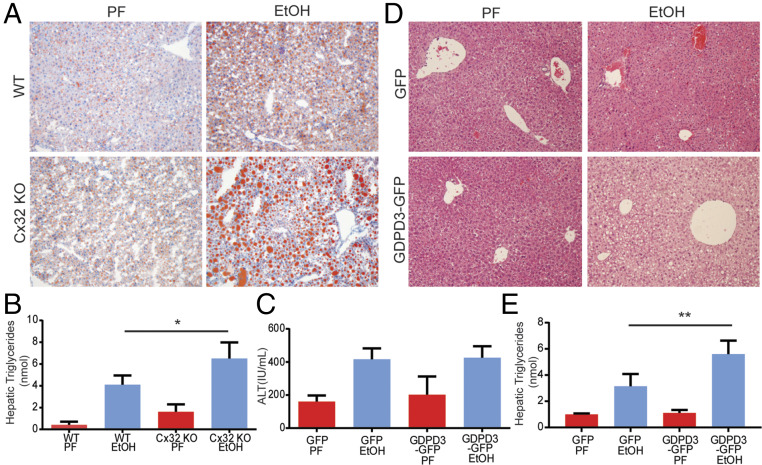
Alcohol-related liver disease (ALD) accounts for the majority of cirrhosis and liver-related deaths worldwide. Activation of IFN-regulatory factor (IRF3) initiates alcohol-induced hepatocyte apoptosis, which fuels a robust secondary inflammatory response that drives ALD. The dominant molecular mechanism by which alcohol activates IRF3 and the pathways that amplify inflammatory signals in ALD remains unknown. Here we show that cytoplasmic sensor cyclic guanosine monophosphate-adenosine monophosphate (AMP) synthase (cGAS) drives IRF3 activation in both alcohol-injured hepatocytes and the neighboring parenchyma via a gap junction intercellular communication pathway. Hepatic RNA-seq analysis of patients with a wide spectrum of ALD revealed that expression of the cGAS-IRF3 pathway correlated positively with disease severity. Alcohol-fed mice demonstrated increased hepatic expression of the cGAS-IRF3 pathway. Mice genetically deficient in cGAS and IRF3 were protected against ALD. Ablation of cGAS in hepatocytes only phenocopied this hepatoprotection, highlighting the critical role of hepatocytes in fueling the cGAS-IRF3 response to alcohol. We identified connexin 32 (Cx32), the predominant hepatic gap junction, as a critical regulator of spreading cGAS-driven IRF3 activation through the liver parenchyma. Disruption of Cx32 in ALD impaired IRF3-stimulated gene expression, resulting in decreased hepatic injury despite an increase in hepatic steatosis. Taken together, these results identify cGAS and Cx32 as key factors in ALD pathogenesis and as potential therapeutic targets for hepatoprotection.
Keywords: IRF3; alcohol liver; cGAS; connexin; innate immunity.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
Competing interest statement: The authors declare a competing interest. K.R.K., M.Y.L., and S.J.P. have equity interest in Heprotech Inc. M.K.G. has equity interest in New Amsterdam Genomics. All other authors have no conflicts to declare.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
