

Amyloid precursor protein processing in human neurons with an allelic series of the PSEN1 intron 4 deletion mutation and total presenilin-1 knockout
- PMID: 32395715
- PMCID: PMC7212081
- DOI: 10.1093/braincomms/fcz024
Amyloid precursor protein processing in human neurons with an allelic series of the PSEN1 intron 4 deletion mutation and total presenilin-1 knockout
Abstract
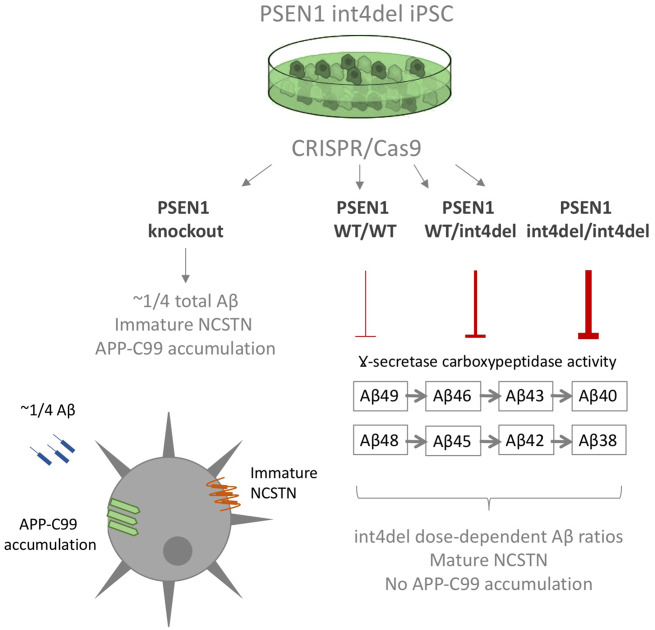
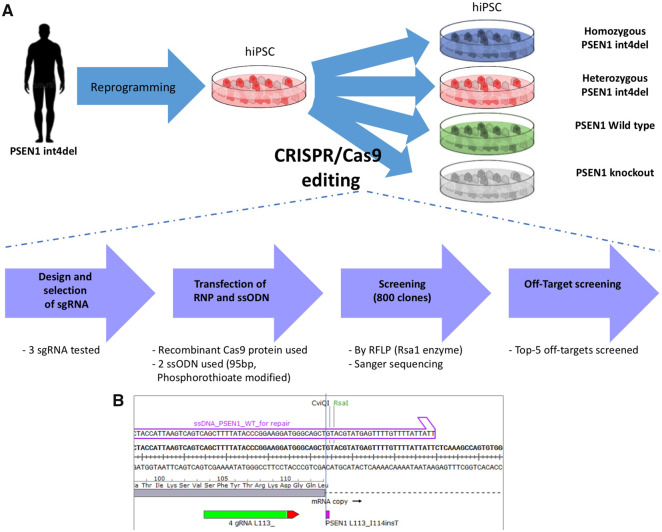
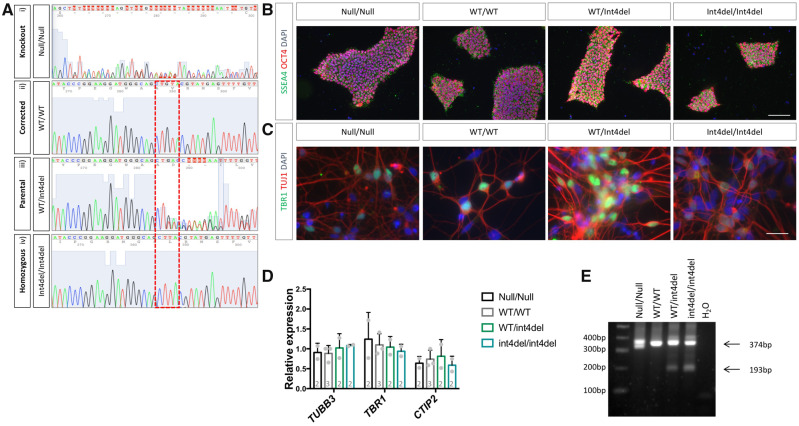
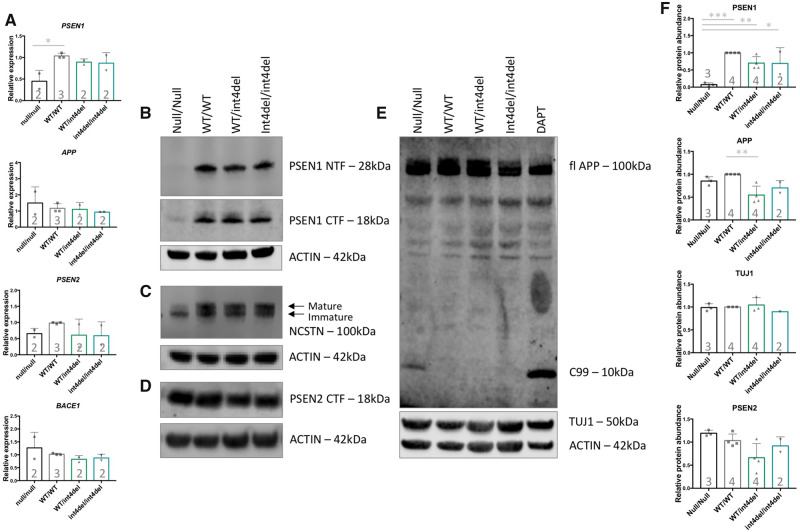
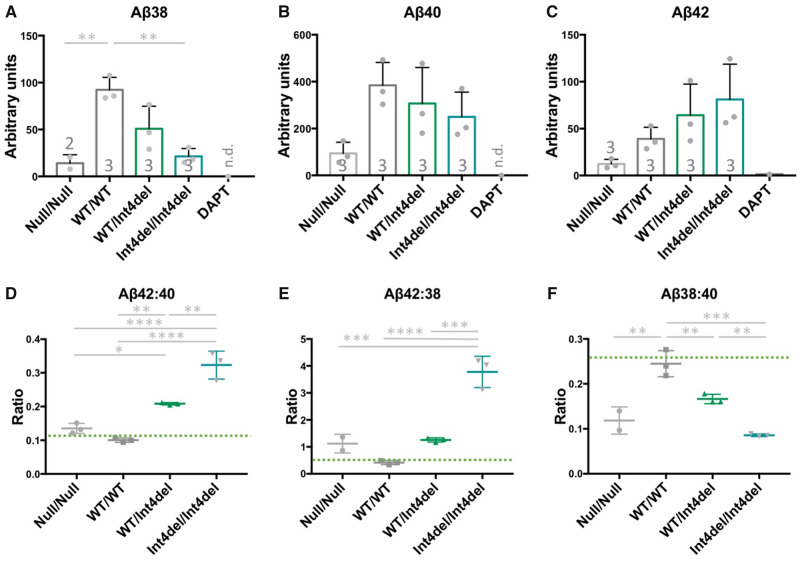
Mutations in presenilin-1 (PSEN1), encoding the catalytic subunit of the amyloid precursor protein-processing enzyme γ-secretase, cause familial Alzheimer's disease. However, the mechanism of disease is yet to be fully understood and it remains contentious whether mutations exert their effects predominantly through gain or loss of function. To address this question, we generated an isogenic allelic series for the PSEN1 mutation intron 4 deletion; represented by control, heterozygous and homozygous mutant induced pluripotent stem cells in addition to a presenilin-1 knockout line. Induced pluripotent stem cell-derived cortical neurons reveal reduced, yet detectable amyloid-beta levels in the presenilin-1 knockout line, and a mutant gene dosage-dependent defect in amyloid precursor protein processing in PSEN1 intron 4 deletion lines, consistent with reduced processivity of γ-secretase. The different effects of presenilin-1 knockout and the PSEN1 intron 4 deletion mutation on amyloid precursor protein-C99 fragment accumulation, nicastrin maturation and amyloid-beta peptide generation support distinct consequences of familial Alzheimer's diseaseassociated mutations and knockout of presenilin-1 on the function of γ-secretase.
Keywords: amyloid beta; Alzheimer’s disease; CRISPR/Cas9; iPSCs.
Conflict of interest statement
Competing interests The authors report no competing interests.
Figures
References
-
- De Jonghe C, Cruts M, Rogaeva EA, Tysoe C, Singleton A, Vanderstichele H, et al.Aberrant splicing in the presenilin-1 intron 4 mutation causes presenile Alzheimer’s disease by increased Abeta42 secretion. Hum Mol Genet 1999; 8: 1529–40. - PubMed
-
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, et al.Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998; 391: 387. - PubMed
-
- De Strooper B. Aph-1, Pen-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron 2003; 38: 9.. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources