

Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations
- PMID: 32396767
- PMCID: PMC7284146
- DOI: 10.1080/07391102.2020.1768149
Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations
Abstract
The recent outbreak of novel "coronavirus disease 2019" (COVID-19) has spread rapidly worldwide, causing a global pandemic. In the present work, we have elucidated the mechanism of binding of two inhibitors, namely α-ketoamide and Z31792168, to SARS-CoV-2 main protease (Mpro or 3CLpro) by using all-atom molecular dynamics simulations and free energy calculations. We calculated the total binding free energy (ΔGbind) of both inhibitors and further decomposed ΔGbind into various forces governing the complex formation using the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method. Our calculations reveal that α-ketoamide is more potent (ΔGbind= - 9.05 kcal/mol) compared to Z31792168 (ΔGbind= - 3.25 kcal/mol) against COVID-19 3CLpro. The increase in ΔGbind for α-ketoamide relative to Z31792168 arises due to an increase in the favorable electrostatic and van der Waals interactions between the inhibitor and 3CLpro. Further, we have identified important residues controlling the 3CLpro-ligand binding from per-residue based decomposition of the binding free energy. Finally, we have compared ΔGbind of these two inhibitors with the anti-HIV retroviral drugs, such as lopinavir and darunavir. It is observed that α-ketoamide is more potent compared to lopinavir and darunavir. In the case of lopinavir, a decrease in van der Waals interactions is responsible for the lower binding affinity compared to α-ketoamide. On the other hand, in the case of darunavir, a decrease in the favorable intermolecular electrostatic and van der Waals interactions contributes to lower affinity compared to α-ketoamide. Our study might help in designing rational anti-coronaviral drugs targeting the SARS-CoV-2 main protease.Communicated by Ramaswamy H. Sarma.
Keywords: COVID-19; MM-PBSA; Molecular Dynamics; SARS-CoV-2 3CLpro; binding free energy.
Figures
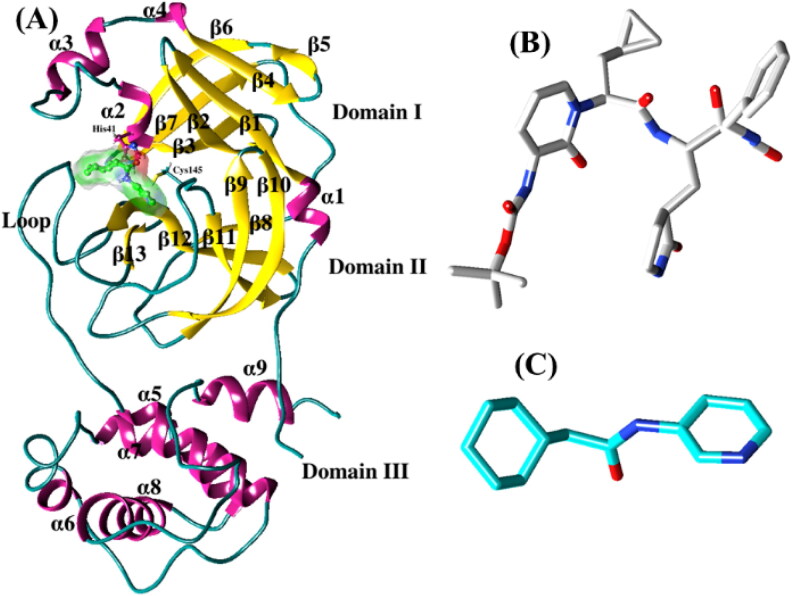
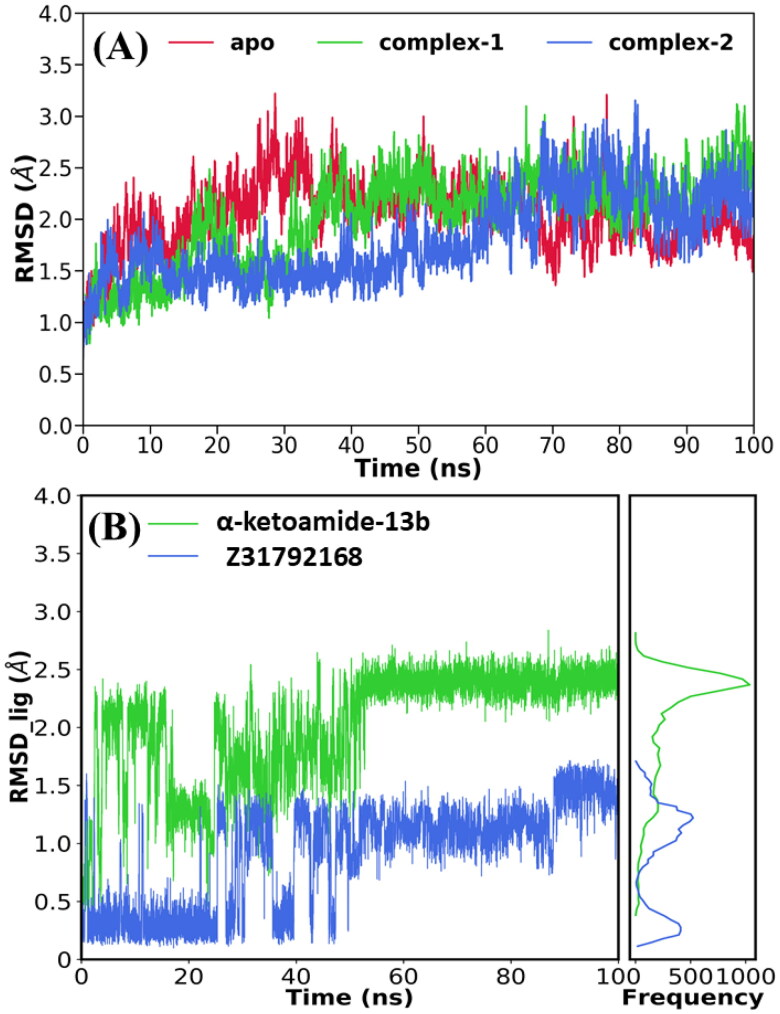
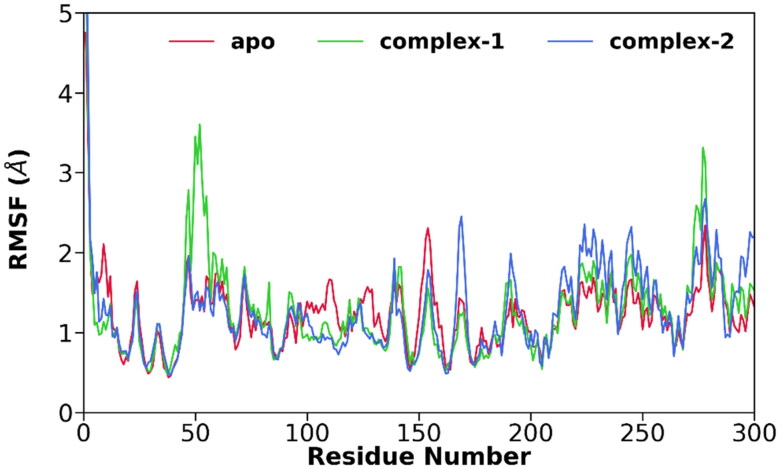
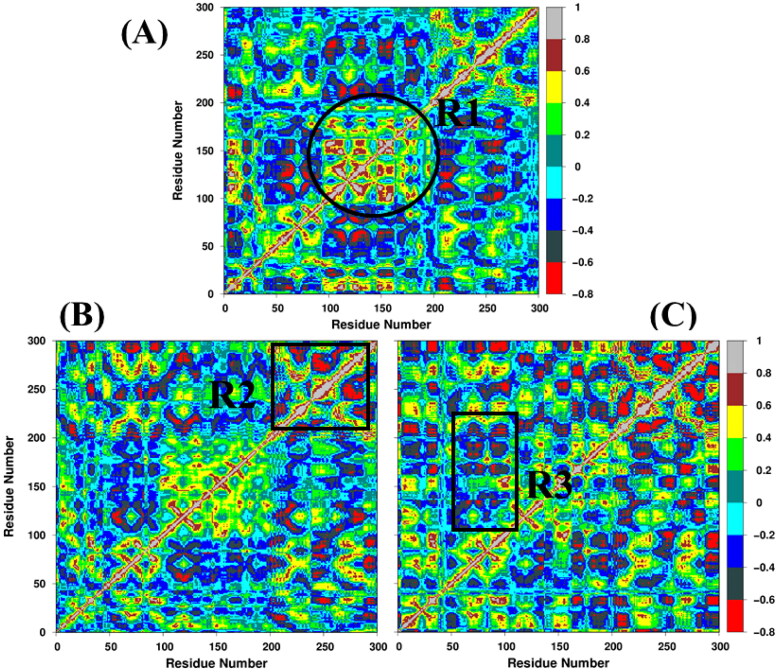
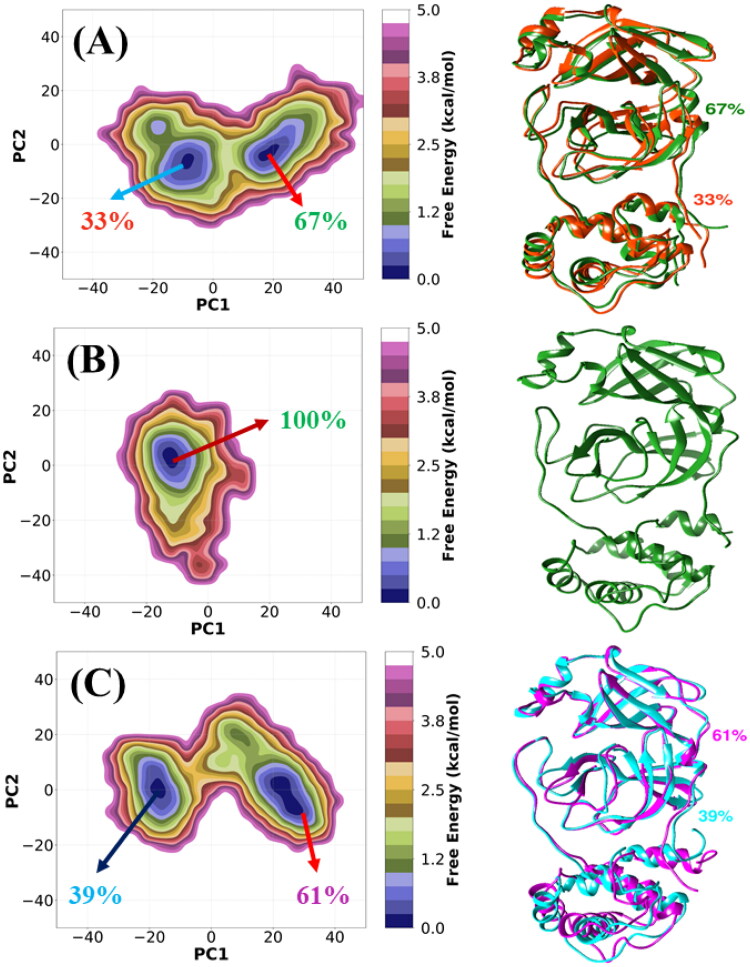
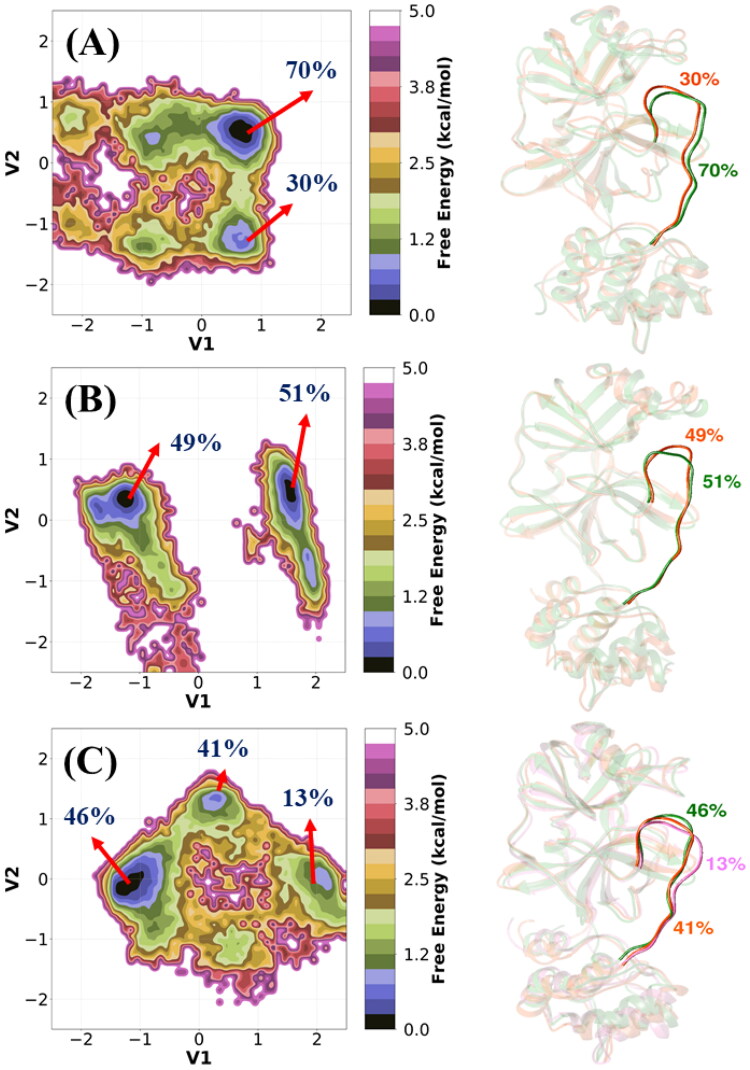
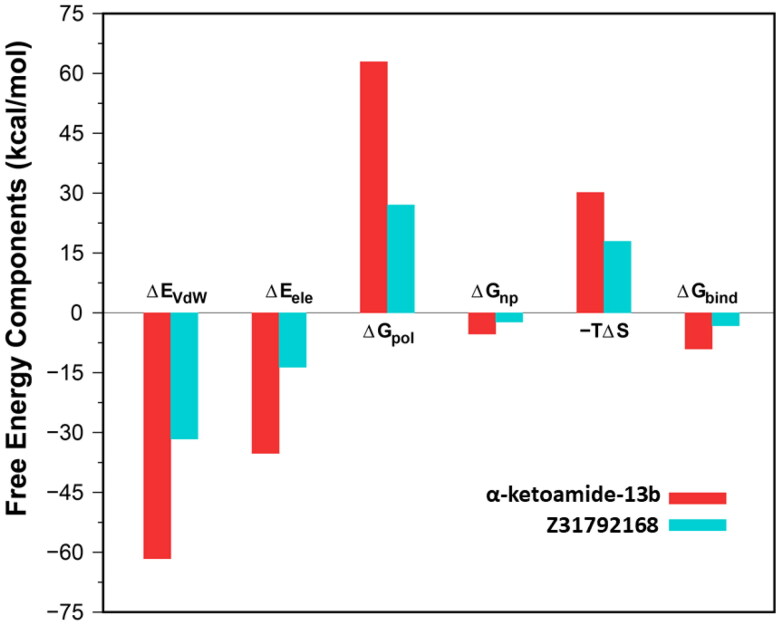
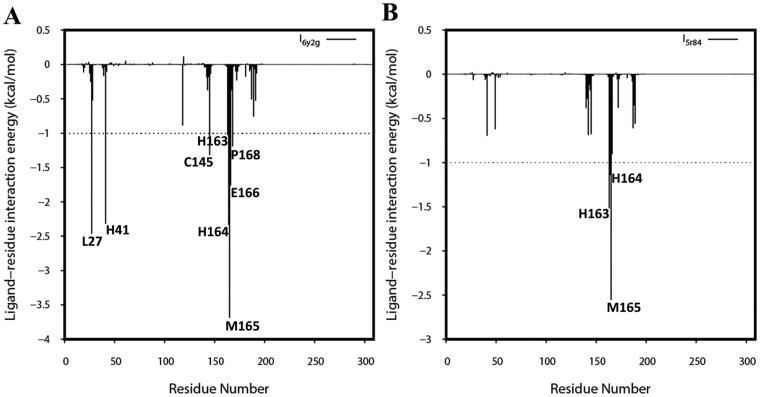
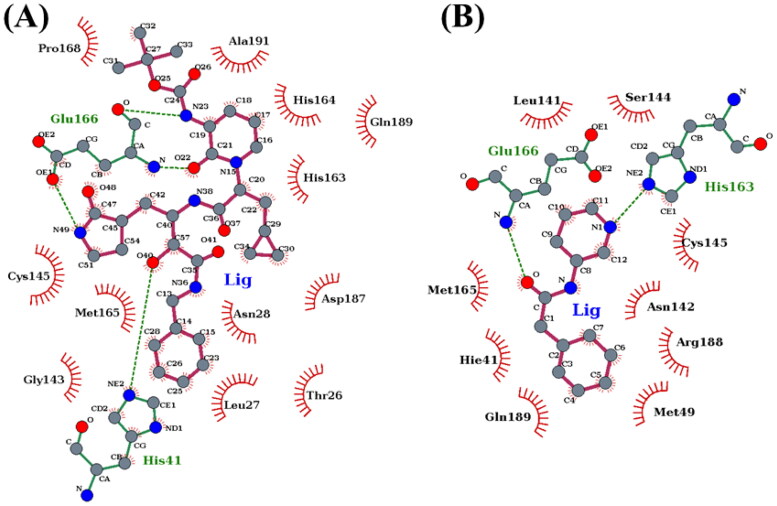
References
-
- Abdelli I., Hassani F., Bekkel Brikci S., & Ghalem S. (2020). In silico study the inhibition of Angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from western Algeria. Journal of Biomolecular Structure and Dynamics. 10.1080/07391102.07392020.01763199 - DOI - PMC - PubMed
-
- Berendsen H. J., Postma J. v., van Gunsteren W. F., DiNola A., & Haak J. (1984). Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics, 81(8), 3684–3690. 10.1063/1.448118 - DOI
-
- Berman H. M., Battistuz T., Bhat T. N., Bluhm W. F., Bourne P. E., Burkhardt K., Feng Z., Gilliland G. L., Iype L., Jain S., Fagan P., Marvin J., Padilla D., Ravichandran V., Schneider B., Thanki N., Weissig H., Westbrook J. D., & Zardecki C. (2002). The protein data bank. Acta Crystallographica. Section D, Biological Crystallography, 58(Pt 6 No 1), 899–907. 10.1107/s0907444902003451 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous