

Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates
- PMID: 32397599
- PMCID: PMC7247013
- DOI: 10.3390/ijms21093369
Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates
Abstract
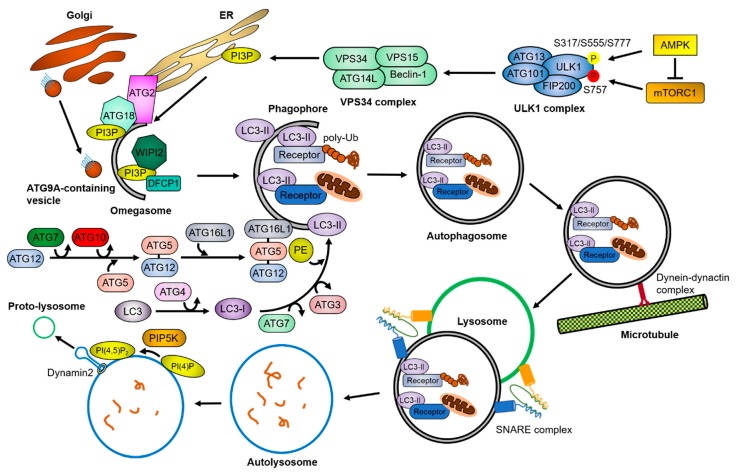
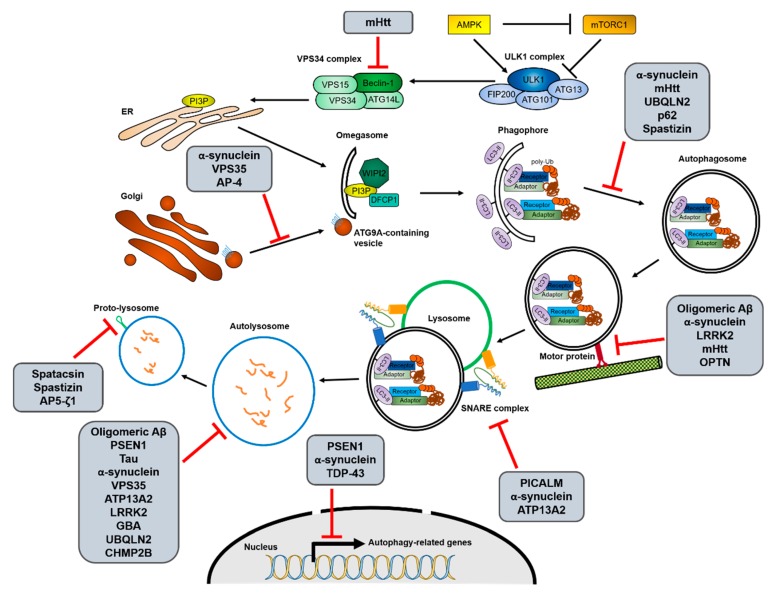
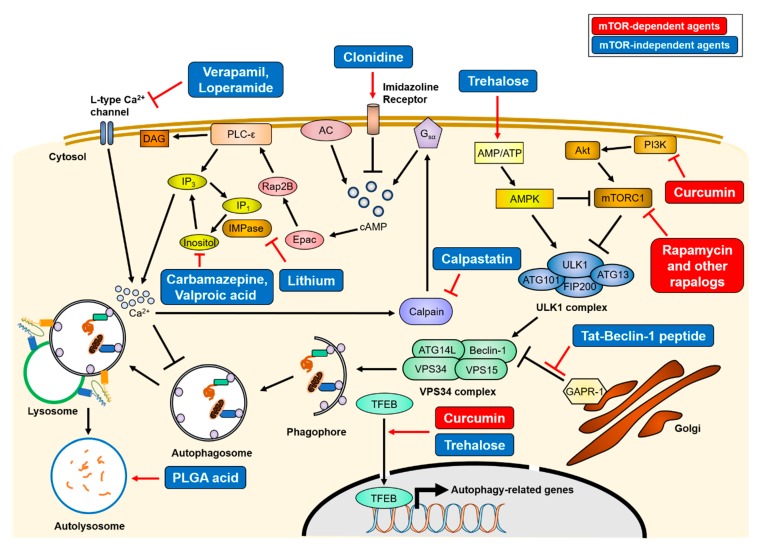
Cells have developed elaborate quality-control mechanisms for proteins and organelles to maintain cellular homeostasis. Such quality-control mechanisms are maintained by conformational folding via molecular chaperones and by degradation through the ubiquitin-proteasome or autophagy-lysosome system. Accumulating evidence suggests that impaired autophagy contributes to the accumulation of intracellular inclusion bodies consisting of misfolded proteins, which is a hallmark of most neurodegenerative diseases. In addition, genetic mutations in core autophagy-related genes have been reported to be linked to neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and Huntington's disease. Conversely, the pathogenic proteins, such as amyloid β and α-synuclein, are detrimental to the autophagy pathway. Here, we review the recent advances in understanding the relationship between autophagic defects and the pathogenesis of neurodegenerative diseases and suggest autophagy induction as a promising strategy for the treatment of these conditions.
Keywords: autophagy; neurodegenerative disease; protein aggregates.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Di Nardo A., Wertz M.H., Kwiatkowski E., Tsai P.T., Leech J.D., Greene-Colozzi E., Goto J., Dilsiz P., Talos D.M., Clish C.B., et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum. Mol. Genet. 2014;23:3865–3874. doi: 10.1093/hmg/ddu101. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
