

Conserved Dynamic Mechanism of Allosteric Response to L-arg in Divergent Bacterial Arginine Repressors
- PMID: 32397647
- PMCID: PMC7248756
- DOI: 10.3390/molecules25092247
Conserved Dynamic Mechanism of Allosteric Response to L-arg in Divergent Bacterial Arginine Repressors
Abstract
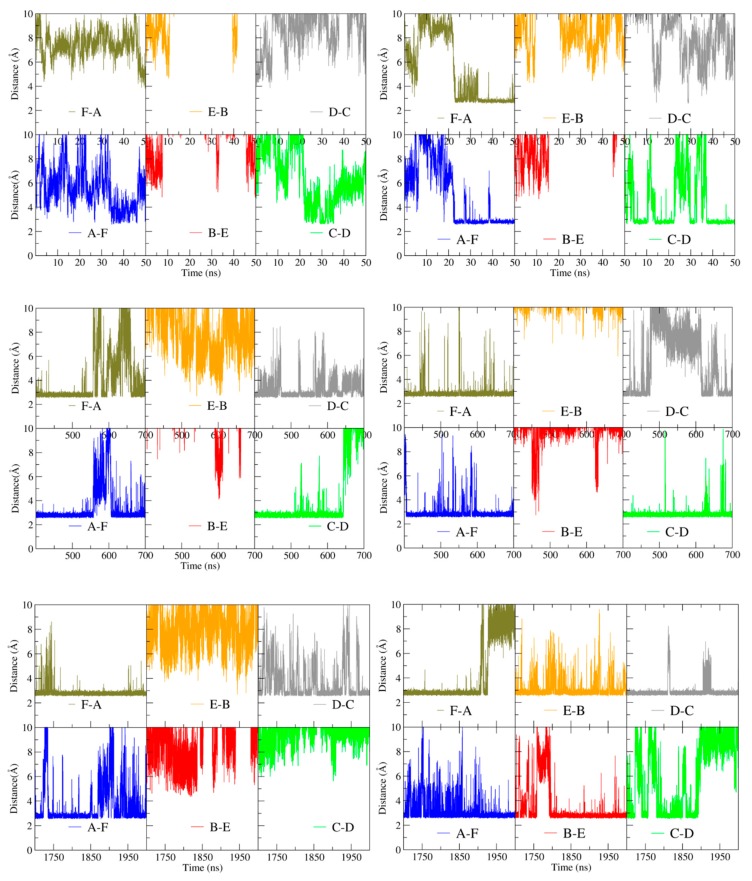
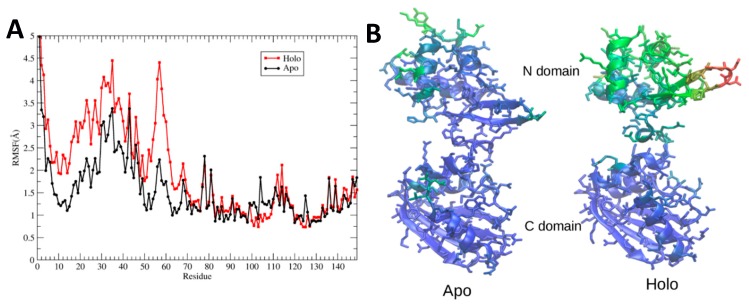
Hexameric arginine repressor, ArgR, is the feedback regulator of bacterial L-arginine regulons, and sensor of L-arg that controls transcription of genes for its synthesis and catabolism. Although ArgR function, as well as its secondary, tertiary, and quaternary structures, is essentially the same in E. coli and B. subtilis, the two proteins differ significantly in sequence, including residues implicated in the response to L-arg. Molecular dynamics simulations are used here to evaluate the behavior of intact B. subtilis ArgR with and without L-arg, and are compared with prior MD results for a domain fragment of E. coli ArgR. Relative to its crystal structure, B. subtilis ArgR in absence of L-arg undergoes a large-scale rotational shift of its trimeric subassemblies that is very similar to that observed in the E. coli protein, but the residues driving rotation have distinct secondary and tertiary structural locations, and a key residue that drives rotation in E. coli is missing in B. subtilis. The similarity of trimer rotation despite different driving residues suggests that a rotational shift between trimers is integral to ArgR function. This conclusion is supported by phylogenetic analysis of distant ArgR homologs reported here that indicates at least three major groups characterized by distinct sequence motifs but predicted to undergo a common rotational transition. The dynamic consequences of L-arg binding for transcriptional activation of intact ArgR are evaluated here for the first time in two-microsecond simulations of B. subtilis ArgR. L-arg binding to intact B. subtilis ArgR causes a significant further shift in the angle of rotation between trimers that causes the N-terminal DNA-binding domains lose their interactions with the C-terminal domains, and is likely the first step toward adopting DNA-binding-competent conformations. The results aid interpretation of crystal structures of ArgR and ArgR-DNA complexes.
Keywords: entropy; global motion; ligand binding; molecular evolution; salt bridges.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures







References
-
- Ni J., Sakanyan V., Charlier D., Glansdorff N., Van Duyne G.D. Structure of the arginine repressor from Bacillus stearothermophilus. Nat. Struct. Biol. 1999;6:427–432. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
