

Endoplasmic reticulum retention and degradation of a mutation in SLC6A1 associated with epilepsy and autism
- PMID: 32398021
- PMCID: PMC7218610
- DOI: 10.1186/s13041-020-00612-6
Endoplasmic reticulum retention and degradation of a mutation in SLC6A1 associated with epilepsy and autism
Abstract
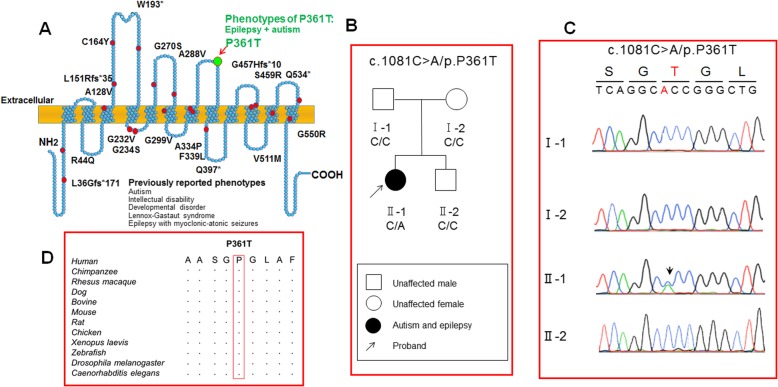
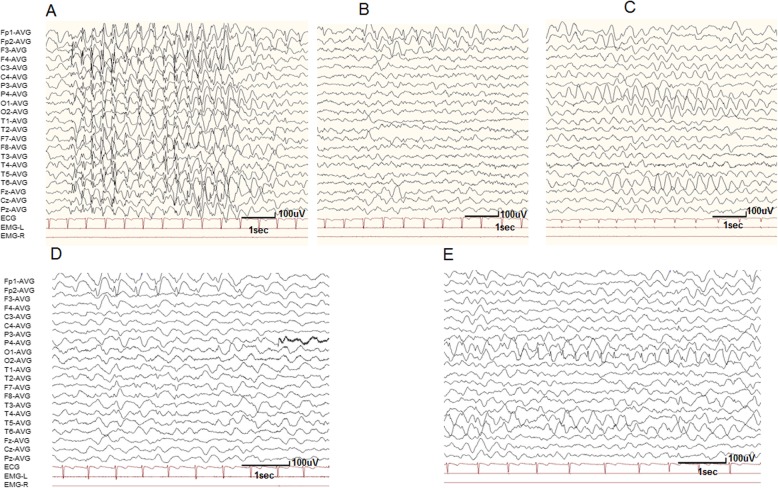
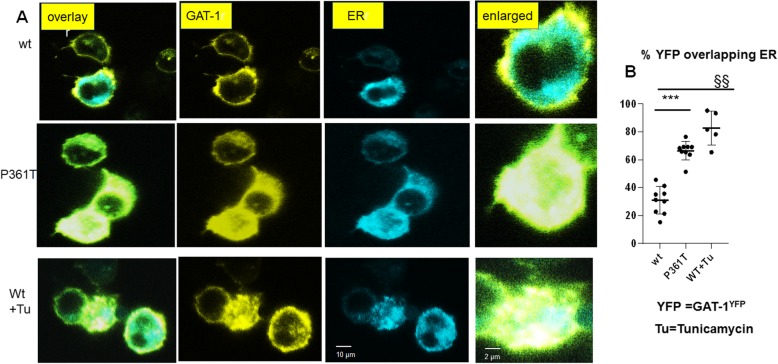
Mutations in SLC6A1, encoding γ-aminobutyric acid (GABA) transporter 1 (GAT-1), have been recently associated with a spectrum of epilepsy syndromes, intellectual disability and autism in clinic. However, the pathophysiology of the gene mutations is far from clear. Here we report a novel SLC6A1 missense mutation in a patient with epilepsy and autism spectrum disorder and characterized the molecular defects of the mutant GAT-1, from transporter protein trafficking to GABA uptake function in heterologous cells and neurons. The heterozygous missense mutation (c1081C to A (P361T)) in SLC6A1 was identified by exome sequencing. We have thoroughly characterized the molecular pathophysiology underlying the clinical phenotypes. We performed EEG recordings and autism diagnostic interview. The patient had neurodevelopmental delay, absence epilepsy, generalized epilepsy, and 2.5-3 Hz generalized spike and slow waves on EEG recordings. The impact of the mutation on GAT-1 function and trafficking was evaluated by 3H GABA uptake, structural simulation with machine learning tools, live cell confocal microscopy and protein expression in mouse neurons and nonneuronal cells. We demonstrated that the GAT-1(P361T) mutation destabilizes the global protein conformation and reduces total protein expression. The mutant transporter protein was localized intracellularly inside the endoplasmic reticulum (ER) with a pattern of expression very similar to the cells treated with tunicamycin, an ER stress inducer. Radioactive 3H-labeled GABA uptake assay indicated the mutation reduced the function of the mutant GAT-1(P361T), to a level that is similar to the cells treated with GAT-1 inhibitors. In summary, this mutation destabilizes the mutant transporter protein, which results in retention of the mutant protein inside cells and reduction of total transporter expression, likely via excessive endoplasmic reticulum associated degradation. This thus likely causes reduced functional transporter number on the cell surface, which then could cause the observed reduced GABA uptake function. Consequently, malfunctioning GABA signaling may cause altered neurodevelopment and neurotransmission, such as enhanced tonic inhibition and altered cell proliferation in vivo. The pathophysiology due to severely impaired GAT-1 function may give rise to a wide spectrum of neurodevelopmental phenotypes including autism and epilepsy.
Keywords: 3H GABA uptake; Autism; Degradation; Endoplasmic reticulum; Epilepsy; GABA transporter 1; Mutation; Protein stability.
Conflict of interest statement
The authors declare that they are no competing interests.
Figures



References
-
- Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli NS, O'Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin TL, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. - DOI - PMC - PubMed
-
- Andang M, Hjerling-Leffler J, Moliner A, Lundgren TK, Castelo-Branco G, Nanou E, Pozas E, Bryja V, Halliez S, Nishimaru H, Wilbertz J, Arenas E, Koltzenburg M, Charnay P, El MA, Ibanez CF, Ernfors P. Histone H2AX-dependent GABA(a) receptor regulation of stem cell proliferation. Nature. 2008;451:460–464. doi: 10.1038/nature06488. - DOI - PubMed
