

Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank
- PMID: 32398145
- PMCID: PMC7218494
- DOI: 10.1186/s13059-020-02023-1
Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank
Abstract
Genomic analyses are sensitive to contamination in public databases caused by incorrectly labeled reference sequences. Here, we describe Conterminator, an efficient method to detect and remove incorrectly labeled sequences by an exhaustive all-against-all sequence comparison. Our analysis reports contamination of 2,161,746, 114,035, and 14,148 sequences in the RefSeq, GenBank, and NR databases, respectively, spanning the whole range from draft to "complete" model organism genomes. Our method scales linearly with input size and can process 3.3 TB in 12 days on a 32-core computer. Conterminator can help ensure the quality of reference databases. Source code (GPLv3): https://github.com/martin-steinegger/conterminator.
Keywords: Contamination; GenBank; Genomes; RefSeq; Software.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
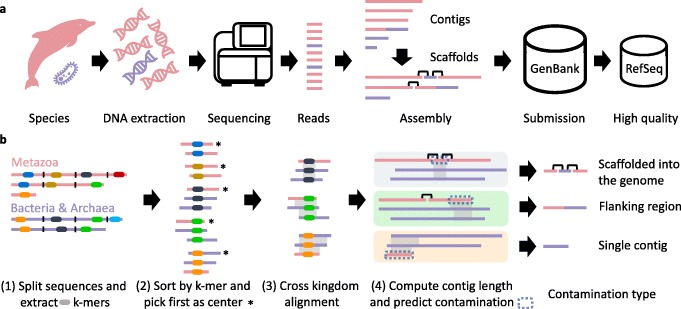
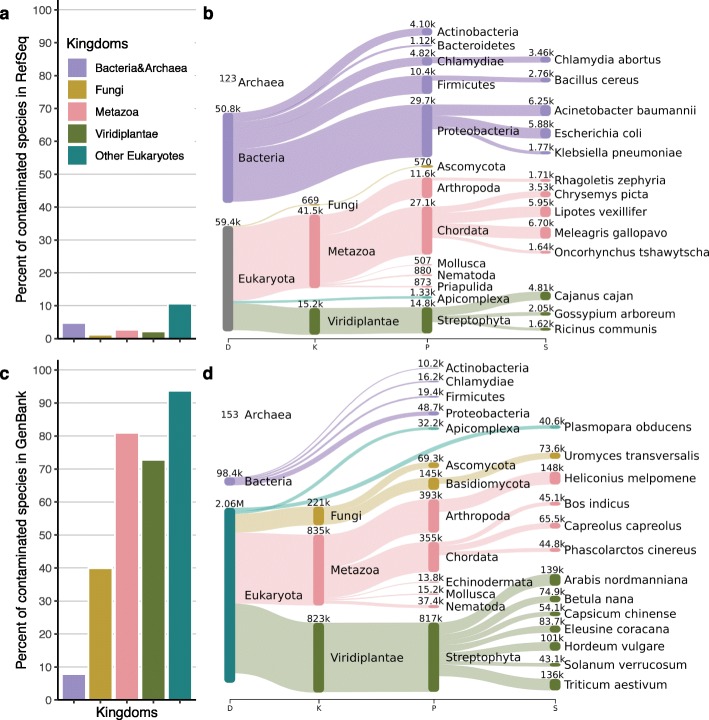
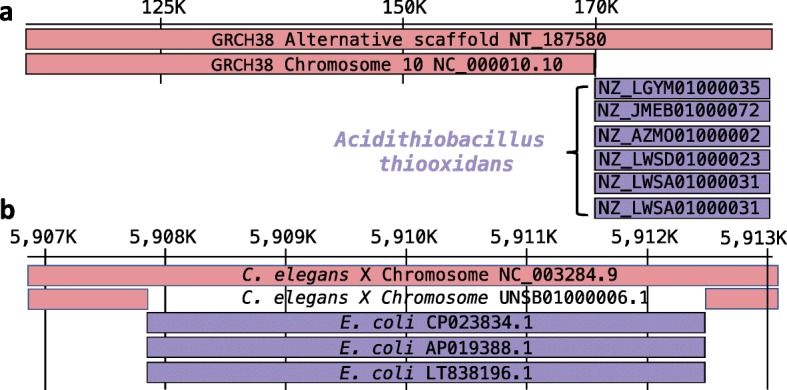
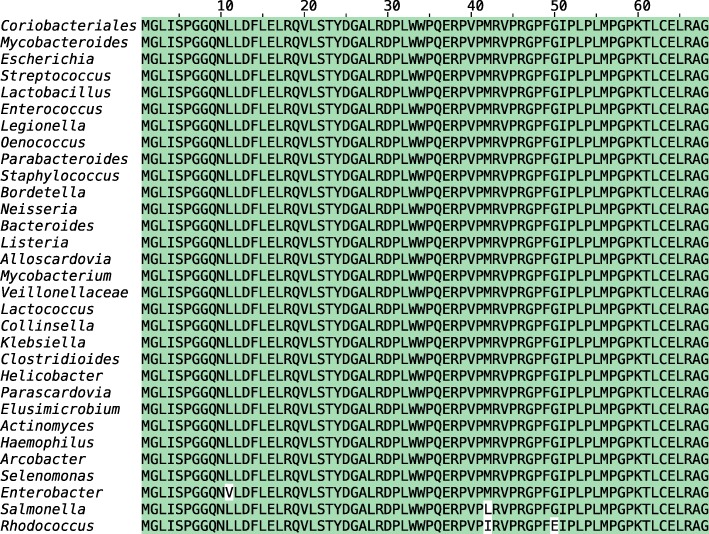
Comment in
-
Contamination in sequence databases.Nat Methods. 2020 Jul;17(7):654. doi: 10.1038/s41592-020-0895-8. Nat Methods. 2020. PMID: 32616930 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
