

Systemic Type I IFN Inflammation in Human ISG15 Deficiency Leads to Necrotizing Skin Lesions
- PMID: 32402279
- PMCID: PMC7331931
- DOI: 10.1016/j.celrep.2020.107633
Systemic Type I IFN Inflammation in Human ISG15 Deficiency Leads to Necrotizing Skin Lesions
Abstract
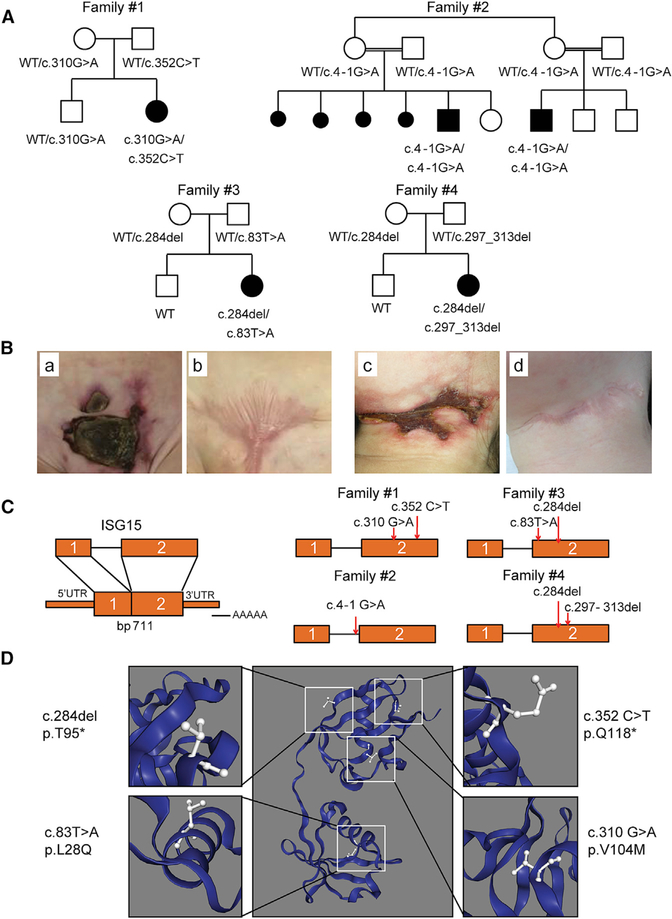
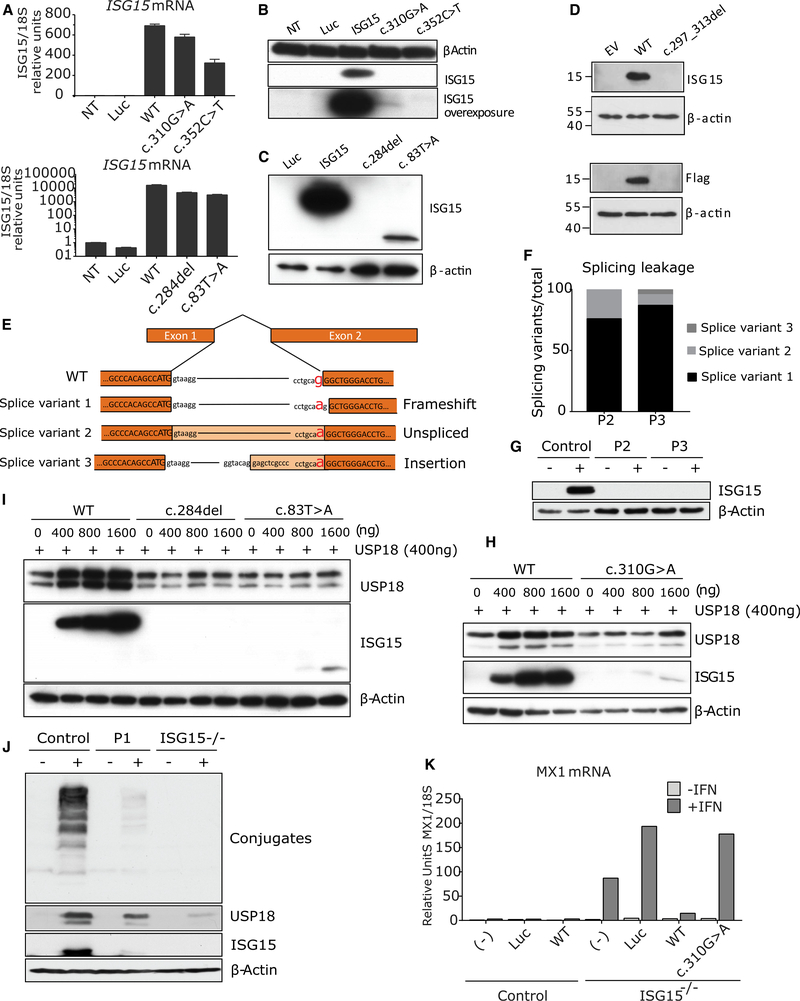
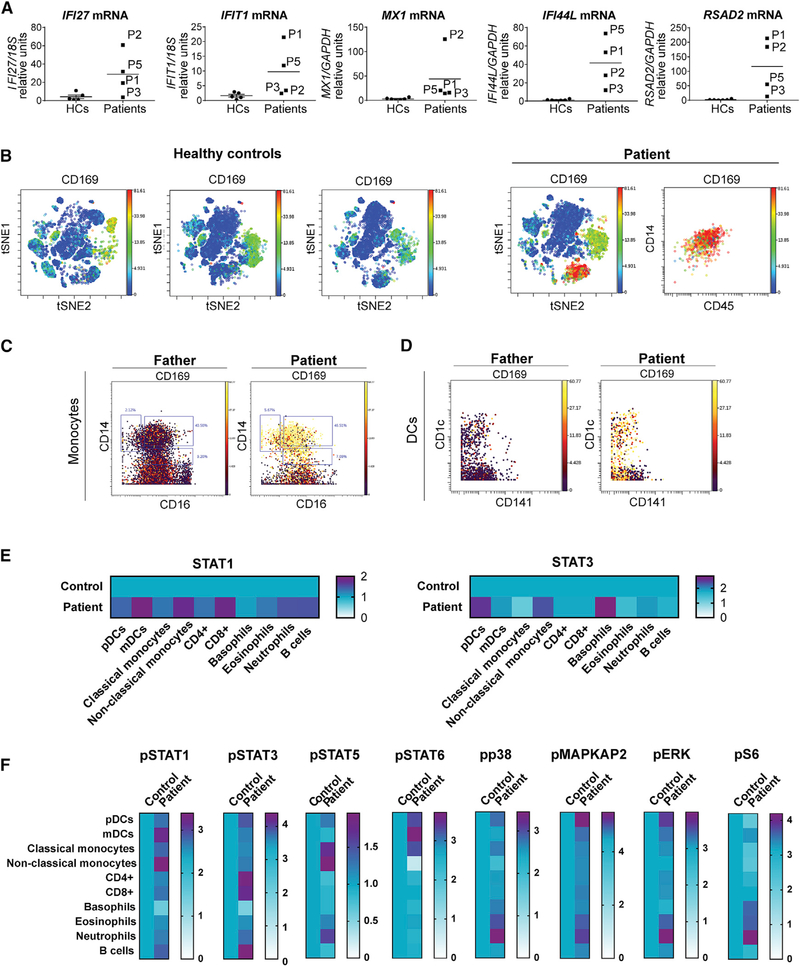
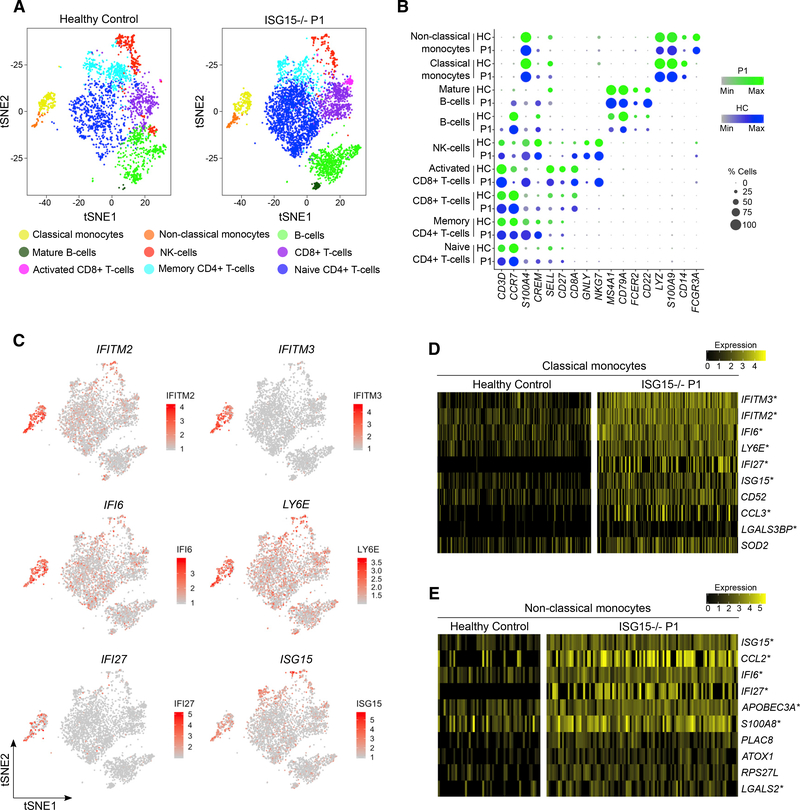
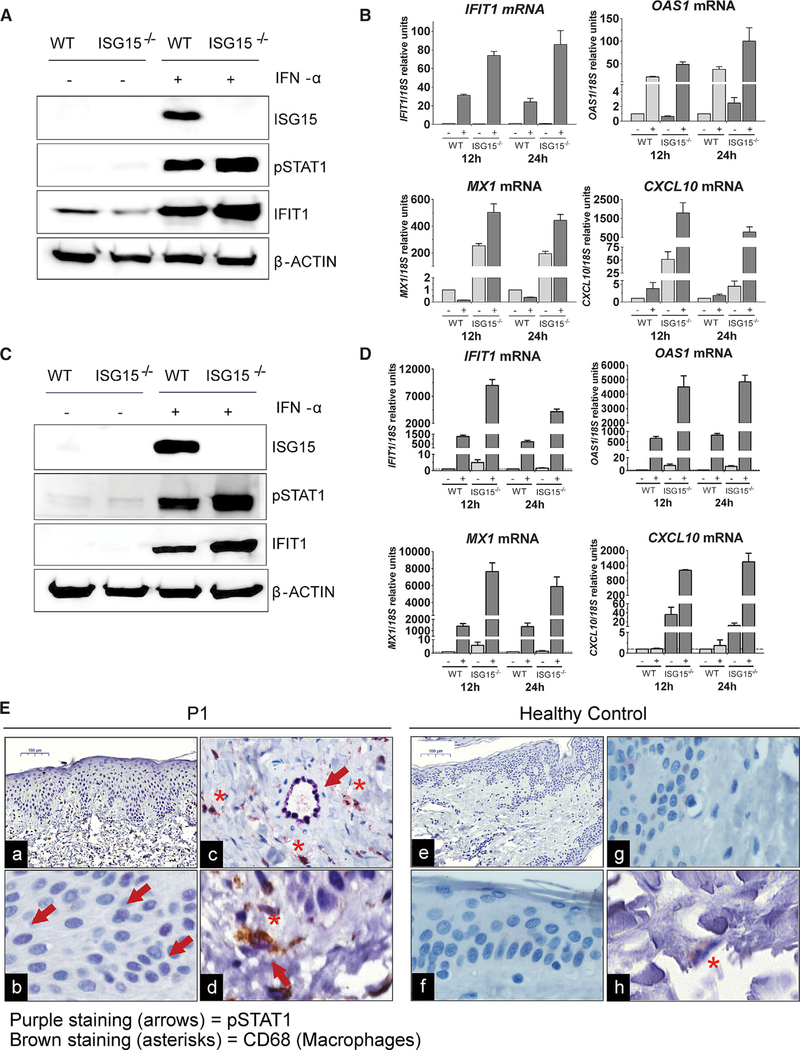
Most monogenic disorders have a primary clinical presentation. Inherited ISG15 deficiency, however, has manifested with two distinct presentations to date: susceptibility to mycobacterial disease and intracranial calcifications from hypomorphic interferon-II (IFN-II) production and excessive IFN-I response, respectively. Accordingly, these patients were managed for their infectious and neurologic complications. Herein, we describe five new patients with six novel ISG15 mutations presenting with skin lesions who were managed for dermatologic disease. Cellularly, we denote striking specificity to the IFN-I response, which was previously assumed to be universal. In peripheral blood, myeloid cells display the most robust IFN-I signatures. In the affected skin, IFN-I signaling is observed in the keratinocytes of the epidermis, endothelia, and the monocytes and macrophages of the dermis. These findings define the specific cells causing circulating and dermatologic inflammation and expand the clinical spectrum of ISG15 deficiency to dermatologic presentations as a third phenotype co-dominant to the infectious and neurologic manifestations.
Keywords: ISG15; Mendelian susceptibility to mycobacterial disease; USP18; endothelial cells; inborn errors of immunity; keratinocytes; myeloid cells; skin inflammation; type I interferonopathy.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Broderick L (2019). Hereditary autoinflammatory disorders: recognition and treatment. Immunol. Allergy Clin. North Am. 39, 13–29. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
