

Proton-Electron Transfer to the Active Site Is Essential for the Reaction Mechanism of Soluble Δ9-Desaturase
- PMID: 32406236
- PMCID: PMC7316153
- DOI: 10.1021/jacs.0c01786
Proton-Electron Transfer to the Active Site Is Essential for the Reaction Mechanism of Soluble Δ9-Desaturase
Abstract
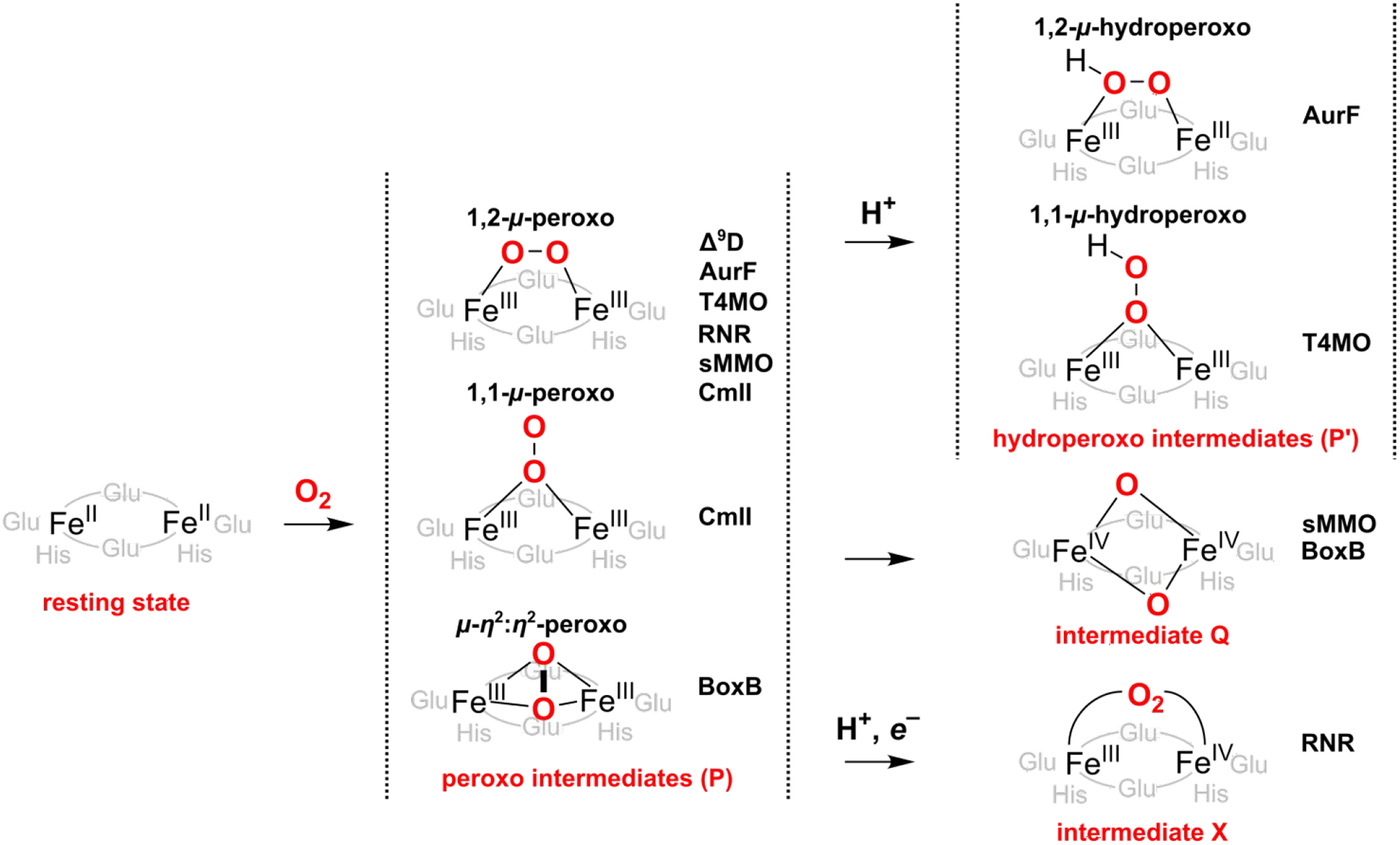
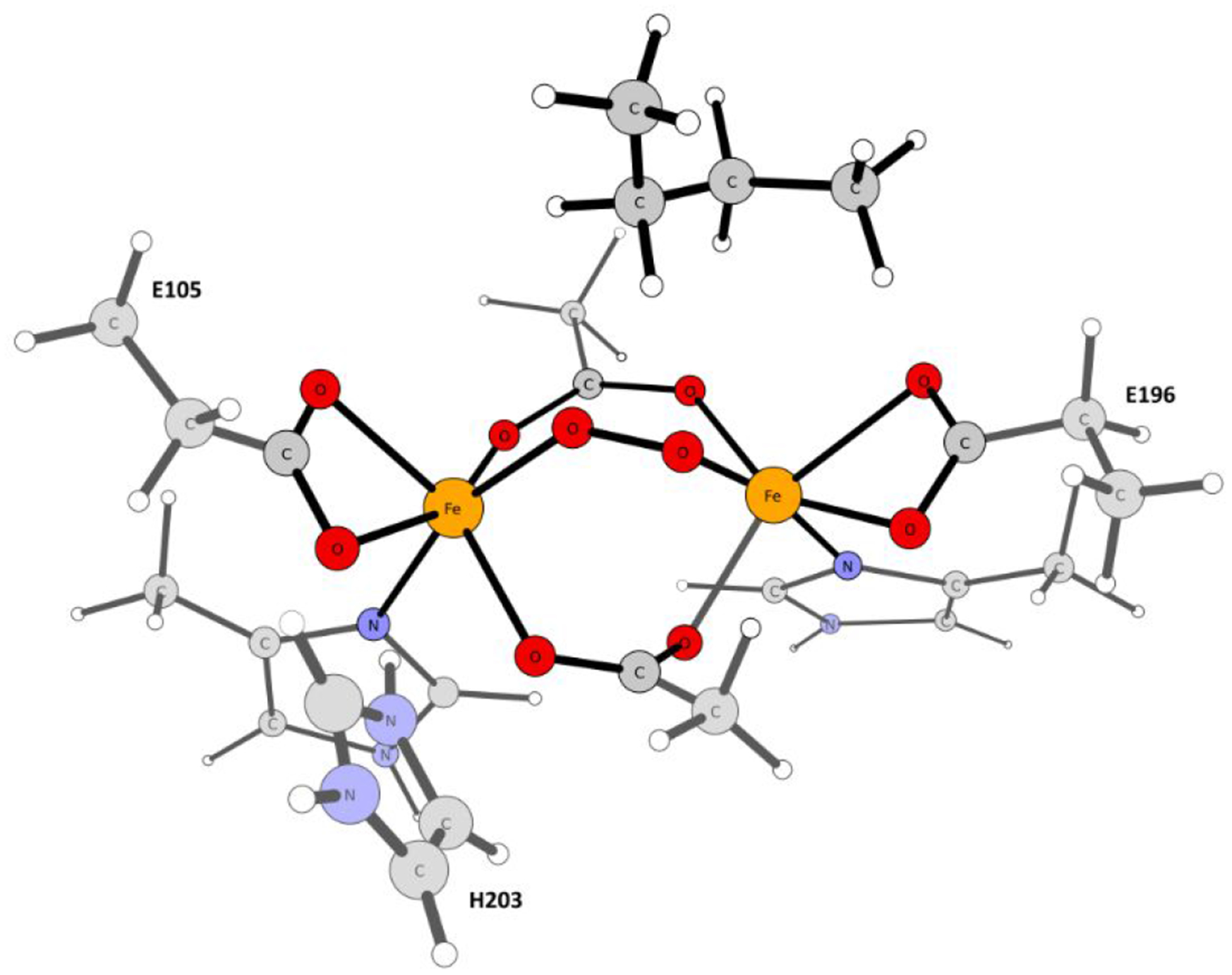
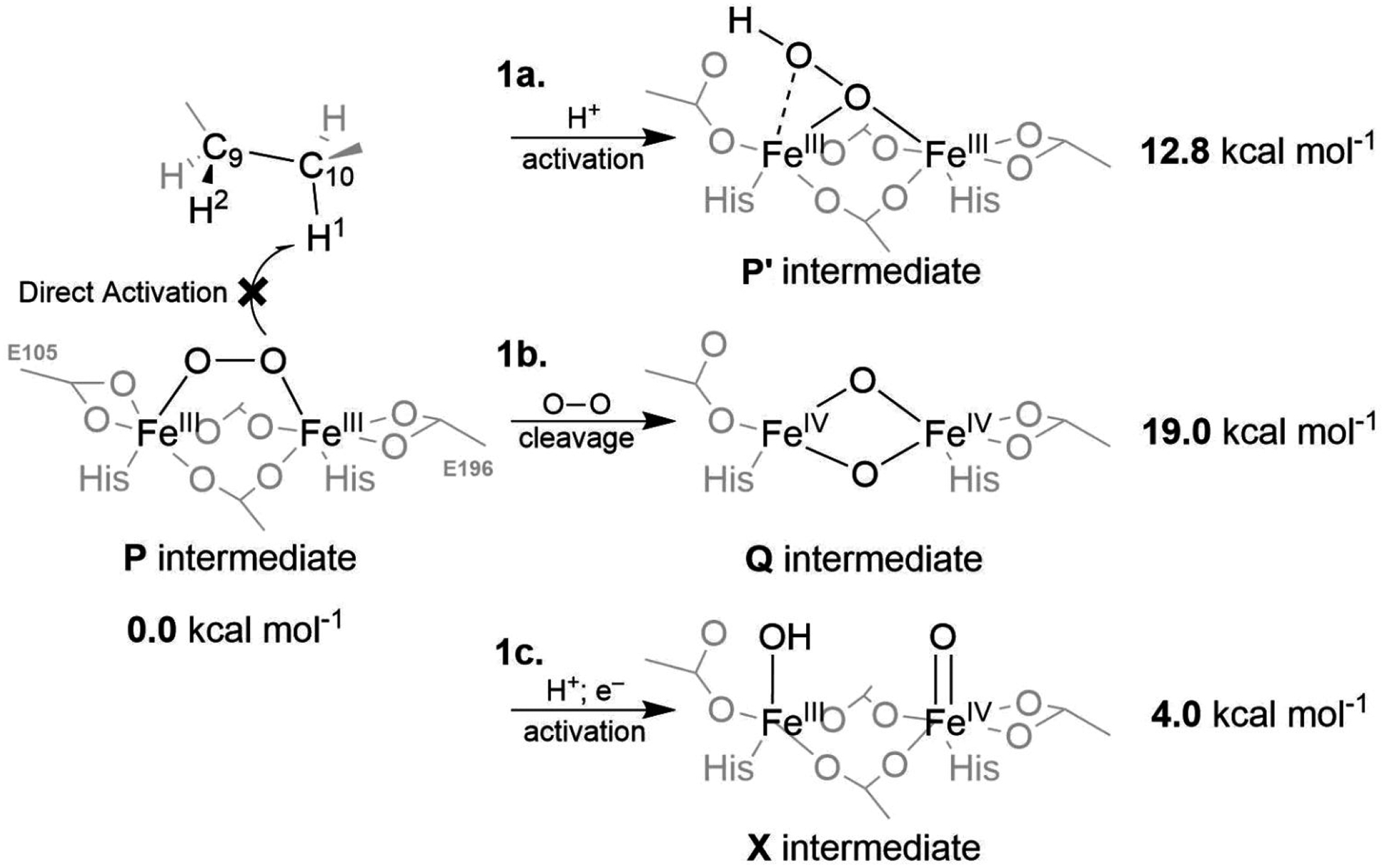
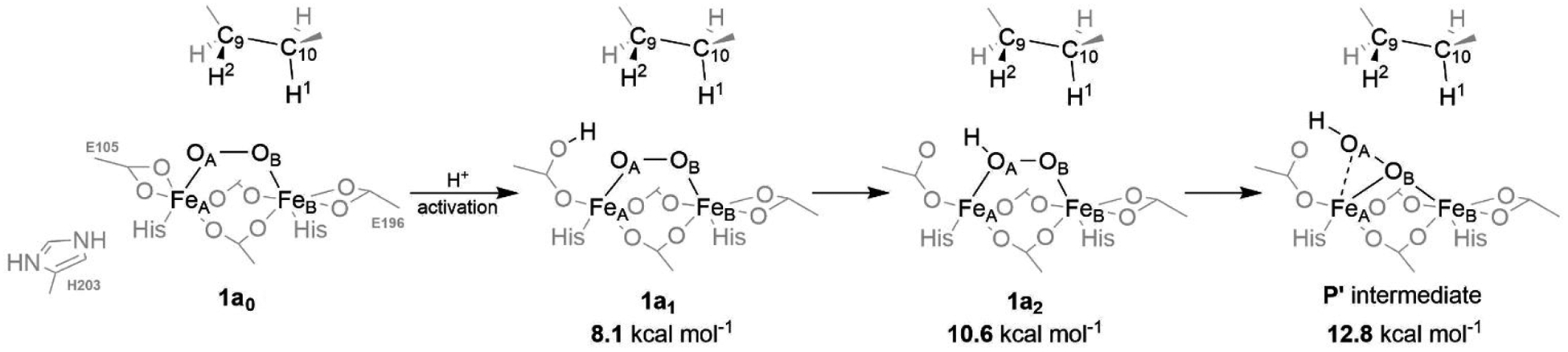
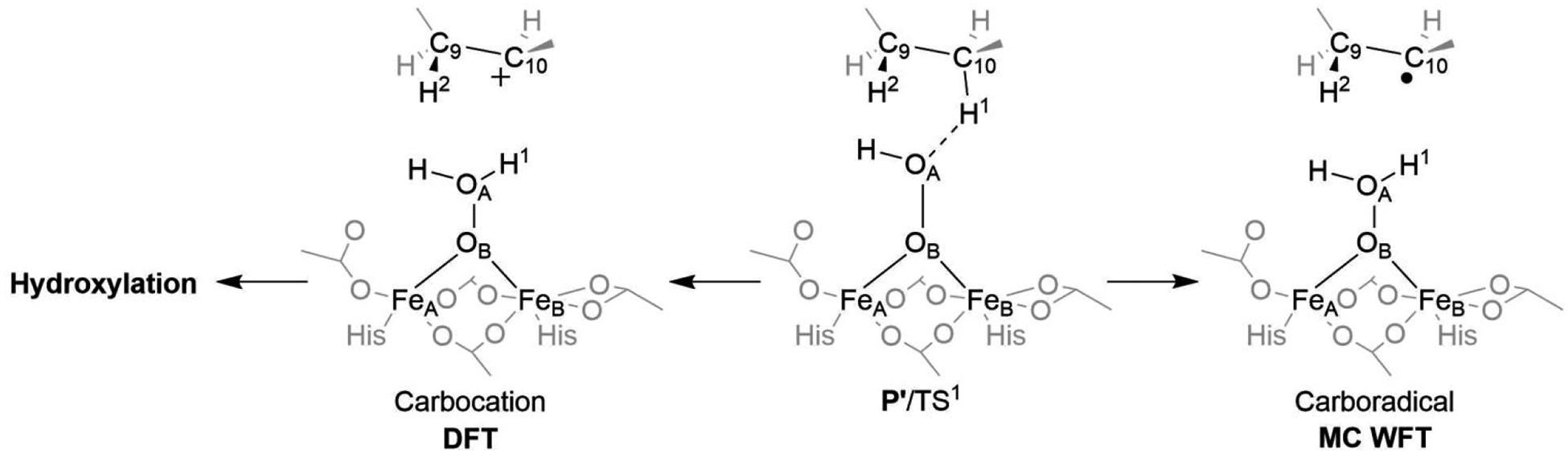
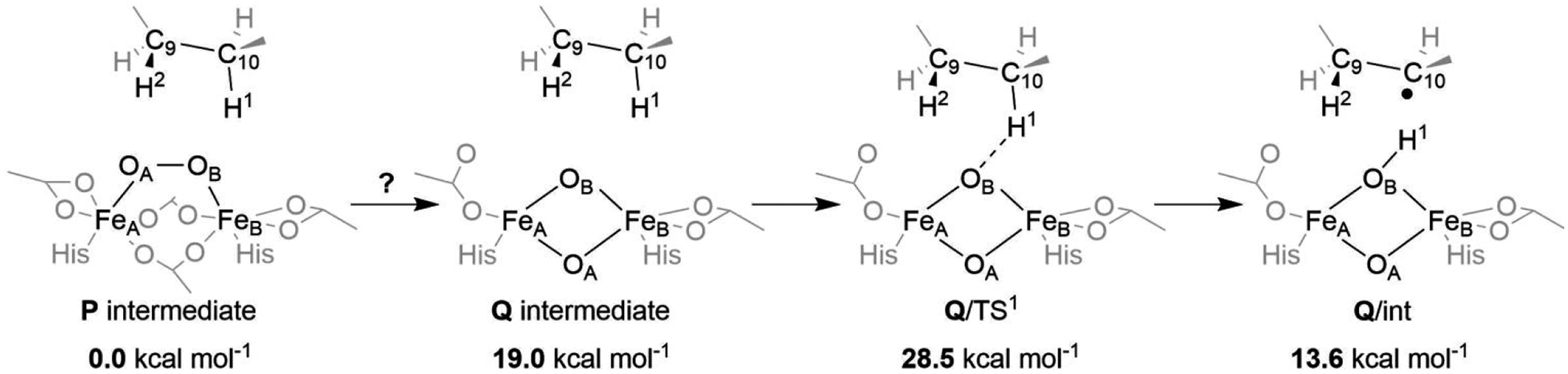
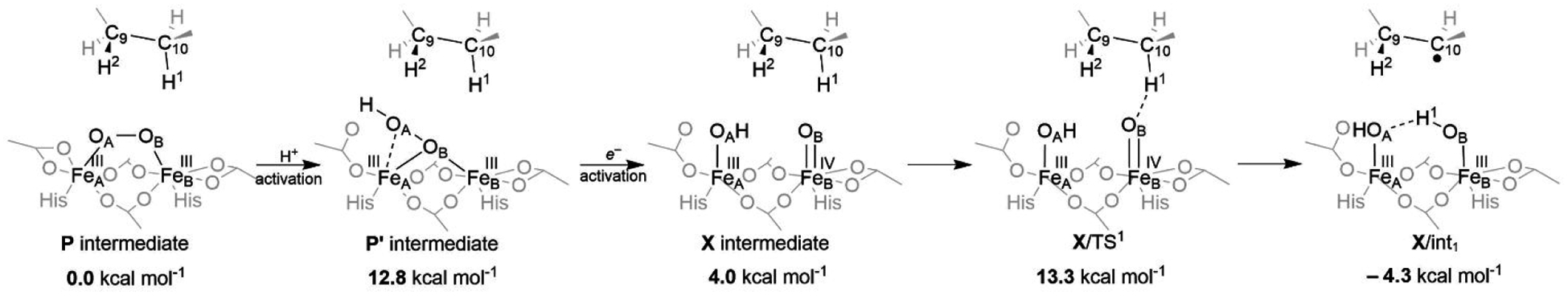
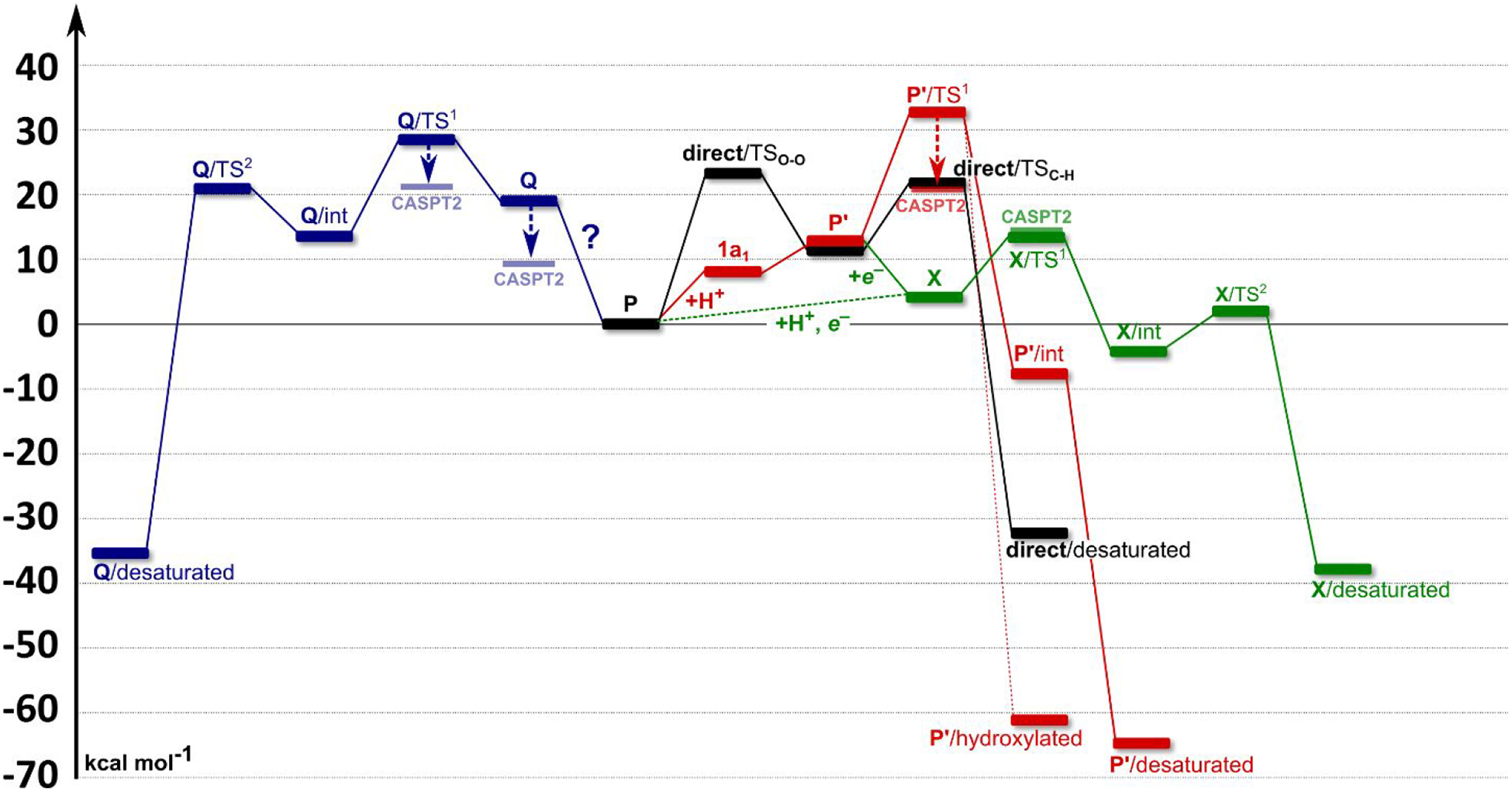
A full understanding of the catalytic action of non-heme iron (NHFe) and non-heme diiron (NHFe2) enzymes is still beyond the grasp of contemporary computational and experimental techniques. Many of these enzymes exhibit fascinating chemo-, regio-, and stereoselectivity, in spite of employing highly reactive intermediates which are necessary for activations of most stable chemical bonds. Herein, we study in detail one intriguing representative of the NHFe2 family of enzymes: soluble Δ9 desaturase (Δ9D), which desaturates rather than performing the thermodynamically favorable hydroxylation of substrate. Its catalytic mechanism has been explored in great detail by using QM(DFT)/MM and multireference wave function methods. Starting from the spectroscopically observed 1,2-μ-peroxo diferric P intermediate, the proton-electron uptake by P is the favored mechanism for catalytic activation, since it allows a significant reduction of the barrier of the initial (and rate-determining) H-atom abstraction from the stearoyl substrate as compared to the "proton-only activated" pathway. Also, we ruled out that a Q-like intermediate (high-valent diamond-core bis-μ-oxo-[FeIV]2 unit) is involved in the reaction mechanism. Our mechanistic picture is consistent with the experimental data available for Δ9D and satisfies fairly stringent conditions required by Nature: the chemo-, stereo-, and regioselectivity of the desaturation of stearic acid. Finally, the mechanisms evaluated are placed into a broader context of NHFe2 chemistry, provided by an amino acid sequence analysis through the families of the NHFe2 enzymes. Our study thus represents an important contribution toward understanding the catalytic action of the NHFe2 enzymes and may inspire further work in NHFe(2) biomimetic chemistry.
Figures
Similar articles
-
Understanding desaturation/hydroxylation activity of castor stearoyl Δ9-Desaturase through rational mutagenesis.Comput Struct Biotechnol J. 2022 Mar 14;20:1378-1388. doi: 10.1016/j.csbj.2022.03.010. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 35386101 Free PMC article.
-
Reactivity of the binuclear non-heme iron active site of Δ⁹ desaturase studied by large-scale multireference ab initio calculations.J Am Chem Soc. 2014 Nov 12;136(45):15977-91. doi: 10.1021/ja506934k. Epub 2014 Oct 31. J Am Chem Soc. 2014. PMID: 25313991
-
Structural and spectroscopic properties of the peroxodiferric intermediate of Ricinus communis soluble Δ9 desaturase.Inorg Chem. 2012 Mar 5;51(5):2806-20. doi: 10.1021/ic2018067. Epub 2012 Feb 14. Inorg Chem. 2012. PMID: 22332845
-
Mono- and binuclear non-heme iron chemistry from a theoretical perspective.J Biol Inorg Chem. 2016 Sep;21(5-6):619-44. doi: 10.1007/s00775-016-1357-8. Epub 2016 May 26. J Biol Inorg Chem. 2016. PMID: 27229513 Review.
-
Desaturases: emerging models for understanding functional diversification of diiron-containing enzymes.J Biol Chem. 2009 Jul 10;284(28):18559-63. doi: 10.1074/jbc.R900009200. Epub 2009 Apr 10. J Biol Chem. 2009. PMID: 19363032 Free PMC article. Review.
Cited by
-
Generation of a μ-1,2-hydroperoxo FeIIIFeIII and a μ-1,2-peroxo FeIVFeIII Complex.Nat Commun. 2022 Mar 16;13(1):1376. doi: 10.1038/s41467-022-28894-5. Nat Commun. 2022. PMID: 35296656 Free PMC article.
-
Understanding desaturation/hydroxylation activity of castor stearoyl Δ9-Desaturase through rational mutagenesis.Comput Struct Biotechnol J. 2022 Mar 14;20:1378-1388. doi: 10.1016/j.csbj.2022.03.010. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 35386101 Free PMC article.
-
Discovery of a Druggable, Cryptic Pocket in SARS-CoV-2 nsp16 Using Allosteric Inhibitors.ACS Infect Dis. 2023 Oct 13;9(10):1918-1931. doi: 10.1021/acsinfecdis.3c00203. Epub 2023 Sep 20. ACS Infect Dis. 2023. PMID: 37728236 Free PMC article.
-
Nature of the Reactive Biferric Peroxy Intermediate P' in the Arylamine Oxygenases and Related Binuclear Fe Enzymes.J Am Chem Soc. 2025 Apr 9;147(14):11707-11725. doi: 10.1021/jacs.4c11712. Epub 2025 Apr 1. J Am Chem Soc. 2025. PMID: 40167320 Free PMC article.
-
Comprehensive Mechanistic View of the Hydrolysis of Oxadiazole-Based Inhibitors by Histone Deacetylase 6 (HDAC6).ACS Chem Biol. 2023 Jul 21;18(7):1594-1610. doi: 10.1021/acschembio.3c00212. Epub 2023 Jul 1. ACS Chem Biol. 2023. PMID: 37392419 Free PMC article.
References
-
- Solomon EI; Brunold TC; Davis MI; Kemsley JN; Lee S-K; Lehnert N; Neese F; Skulan AJ; Yang Y-S; Zhou J Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev 2000, 100, 235–350. - PubMed
-
- Rokob TA; Chalupský J; Bím D; Andrikopoulos PC; Srnec M; Rulíšek L Mono- and binuclear non-heme iron chemistry from a theoretical perspective. J. Biol. Inorg. Chem 2016, 21, 619–644. - PubMed
-
- Harvey JN On the accuracy of density functional theory in transition metal chemistry. Annu. Rep. Prog. Chem., Sect. C: Phys. Chem 2006, 102, 203–226.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
